同位素稀释液相色谱串联质谱法同时测定饲料中雌激素类药物
2017-10-23王月吴银良
薛 毅 ,张 王月,吕 燕 ,吴银良 *
(1.中国动物疫病预防控制中心,北京100125;2.宁波市农业科学研究院,浙江宁波 315040)
同位素稀释液相色谱串联质谱法同时测定饲料中雌激素类药物
薛 毅1,张 王月1,吕 燕2,吴银良2*
(1.中国动物疫病预防控制中心,北京100125;2.宁波市农业科学研究院,浙江宁波 315040)
试验旨在建立饲料中8种雌激素类药物的同位素稀释液相色谱串联质谱分析方法。样品用乙腈提取,提取液经氮吹后用乙酸乙酯复溶,再利用碳酸钠溶液进行液液萃取净化,净化后吹干乙酸乙酯层,用40%乙腈溶液溶解残渣,经离心过滤膜后进行液相色谱串联质谱分析。采用Acquity HSS T3色谱柱分离,用水-乙腈作为流动相进行梯度洗脱,电喷雾负离子模式电离,多反应监测模式检测,同位素稀释内标法定量。结果表明:8种雌激素类药物在1.0~200 μg/L范围内线性良好,相关系数均在0.9910~0.9992;在1.0~50.0 μg/kg添加水平范围内,平均添加回收率在97.5%~106%。本方法中8种雌激素类药物在饲料中的检测限为0.15~0.30 μg/kg,定量限为0.50~1.0 μg/kg。该方法能满足饲料中8种雌激素类药物含量分析的要求。
雌激素类药物;饲料;同位素稀释;液相色谱-串联质谱法
雌激素是一类有广泛生物活性的类固醇化合物,在畜牧业生产中主要被用来促进母牛同期排卵以提高发情动物的数量和受胎率。然而,雌激素类药物在畜牧业生产上滥用和违规使用会引起动物性食品中雌激素残留,从而导致一系列的人体健康问题,如儿童性早熟、癌症、男性生殖障碍等[1-2]。因此,我国已颁布相关文件,明确禁止己烯雌酚、己烷雌酚等激素类药物在动物生产中使用。
对于雌激素类药物残留确证分析方法,目前主要有气相色谱-质谱法[3-7]和液相色谱-串联质谱法[8-17],分析对象以生物样品、动物源性的食品居多[4-17],饲料样品相对较少[3,8]。由于雌激素类药物挥发性低,用气相色谱-质谱法分析时需要衍生化,导致前处理步骤多操作繁琐,同时定性时相对液相色谱串联质谱法(LC-MS/MS)易干扰,因此近年来,LC-MS/MS分析技术逐渐成为雌激素类药物检测技术研究的热点。本研究通过前处理条件的优化,建立饲料中8种雌激素类药物的准确、灵敏和快速的LC-MS/MS分析方法。
1 材料与方法
1.1 材料
1.1.1 仪器 超高效液相色谱串联质谱仪(美国Waters公司);Sigma离心机(北京博励行仪器有限公司);Milli-Q超纯水仪(美国Millipore公司)。
1.1.2 标准品 雌三醇(96.5%)、雌酮(98.0%)、炔雌醇(98.0%)、17α-雌二醇(98.0%)、17β-雌二醇(96.0%)、已烯雌酚(98.0%)、已烷雌酚(98.0%)、已二烯雌酚(97.0%)、雌三醇-D3(98.1%)、雌酮-D2(95.0%)、炔雌醇-D4(97.0%)、已烯雌酚-D8(98.9%)、已烷雌酚-D4(98.0%)和已二烯雌酚-D6(98.6%)购于加拿大Toronto Research Chemicals(TRC)公司。17α-雌二醇-D2(98.0%)和17β-雌二醇-D2(98.0%)购于加拿大CDN公司。
1.1.3 标准储备溶液配制 分别准确称取雌三醇、雌酮、炔雌醇、17α-雌二醇、17β-雌二醇、已烯雌酚、已烷雌酚、已二烯雌酚、雌三醇-D3、雌酮 -D2、炔雌醇 -D4、17α-雌二醇 -D2、17β-雌二醇-D2、已烯雌酚-D8、已烷雌酚-D4和已二烯雌酚-D6适量于100 mL棕色容量瓶中,用乙腈定容至刻度,配制各标准品的100 μg/mL的标准储备液-18℃保存,有效期6个月。
1.1.4 主要试剂 乙腈(色谱纯)购于美国Fisher公司;乙酸乙酯(分析纯)和碳酸钠(分析纯)购于上海化学试剂公司;超纯水。
1.2 方法
1.2.1 液相色谱串联质谱条件 色谱柱:UPLC Acquity HSS T3 柱子(2.1 mm×100 mm,1.8 μm);流速:0.40 mL/min;进样量:10 μL;柱温:35℃;流动相:A相 :水,B:乙腈;洗脱采用梯度洗脱,程序详见表1;ESI负离子模式;毛细管电压:2.5 kV;源温:150℃;去溶剂气温度:500℃;脱溶剂流速:1 000 L/h;主要监测离子对详见表2。

表1 梯度条件
1.2.2 样品前处理方法 称取(5±0.05)g样品于50 mL聚四氟乙烯离心管中,准确加入50 μL 500 μg/L的同位素标记混合标准溶液,混合15 s后加入25 mL乙腈,在振荡器上振荡提取30 min,以4 000 r/min离心2 min,收集乙腈提取液于50 mL离心管中,氮吹至近干后加入10 mL乙酸乙酯溶解残渣。静置1 min后移取上述乙酸乙酯溶解液至已加有2 mL 5%碳酸钠溶液的50 mL塑料离心管中,涡旋混合1 min,以5 000 r/min离心3 min,弃去下层水相,再加入2 mL 5%碳酸钠溶液,重复涡旋离心步骤后将乙酸乙酯层移入另一50 mL尖底塑料离心管中,在40℃水浴中用氮气吹干,加入1.0 mL 40%的乙腈溶液,涡旋混合0.5 min,过0.2 μm滤膜后待上机。
1.2.3 标准曲线制备 吸取适量的各雌激素标准溶液用水/乙腈(20:80,V/V)混合溶液进行稀释,配制含内标浓度均为12.5 μg/L的1.0、2.0、5.0、20、50、100 μg/L的系列雌激素标准工作溶液,进行LC-MS/MS分析,每一浓度进样3次,以目标物的浓度为横坐标,目标物的定量离子对峰面积与内标离子对峰面积比值为纵坐标,绘制各雌激素标准曲线。
1.2.4 添加回收率试验 分别在各个空白样品中添加100 μL的标准溶液(浓度分别为0.05 μg/L、0.25 mg/L、2.5 mg/L)后样品涡动0.5 min,混匀,静置待挥发至干后,按照样品前处理的1.2.2方法进行处理后进LC-MS/MS分析。添加的浓度为1.0、5.0、50 μg/kg。

表2 8种雌激素类药物及内标物质谱条件
2 结果和分析
2.1 仪器条件的优化 对于雌激素类药物的LCMS/MS分析,由于该类药物分子结构中含有酚羟基,通常采用ESI-作为电离模式[8-14]。因此,本研究利用雌激素标准溶液在全扫描负离子电离模式下,优化毛细管电压、锥孔电压、裂解温度、脱溶剂气流速等参数,得到各雌激素类药物最强的分子离子峰。然后在优化的质谱条件下,对选定的母离子进行子离子扫描,优化碰撞能量等参数,最终经实际空白样品测试,选择表1中既无干扰、灵敏度又高的子离子作为相应药物的定性离子和定量离子。由于17α-雌二醇和17β-雌二醇是一对同分异构体,本研究利用Acquity UPLC HSS T3和BEH C18色谱柱考察流动相为乙腈-水时,在同样梯度条件下的分离情况,结果当采用Acquity UPLC HSS T3柱时分离效果相对较好。
2.2 前处理条件的选择和优化 雌激素属于疏水性有机化合物,易溶于大多数有机溶剂,目前常用的提取溶剂主要有乙酸乙酯[11]、乙腈[9]和叔丁基甲醚[8]。考虑到饲料通常蛋白含量比较高且乙腈具有去蛋白作用,本试验采用不同体积乙腈(15、20、25、30、40 mL)按照1.2.2中的方法对猪配合饲料中8种雌激素类药物进行提取效果研究,结果发现8种雌激素类药物的提取回收率从15 mL的72.1%~86.5%增长到25 mL的90.6%~98.1%,而后趋于稳定,因此本试验选择乙腈体积为25 mL。
对于饲料及动物性食品中雌激素类药物分析前处理净化方法主要有液液萃取法、固相萃取法和QuEChERS方法。其中液液萃取法主要利用雌激素类药物结构中含有酚羟基,可在弱碱性条件下用有机溶剂萃取待测物,从而达到净化的目的;因此本试验在乙腈提取液吹干用乙酸乙酯溶解后采用碳酸钠溶液对其进行净化,结果发现饲料样品中8种雌激素类药物出峰时间无杂质峰干扰,且净化步骤所需时间只有10 min左右,比常规固相萃取方法(通常需要1 h左右)要节约时间80%以上。
2.3 线性试验和基质效应 用流动相配制1.2.3中的浓度系列标准溶液进行LC-MS/MS分析,以标准溶液浓度为横坐标,定量离子对峰面积和内标离子对峰面积的比值为纵坐标,绘制标准曲线。从表3中可知,8种雌激素类药物在1.0~200 μg/L范围内线性相关系数均大于0.990,线性关系良好。同时利用空白样品配制基质标准溶液进行进样分析,分别对系列标准溶液和基质标准溶液以标样浓度为横坐标,定量离子对峰面积为纵坐标进行线性回归分析,计算基质匹配标准曲线和溶剂标准曲线斜率比值,得到基质效应的强弱。在本研究中,猪配合饲料、鸡配合饲料对8种雌激素类药物均产生了不同程度的基质抑制效应(比值介于0.23~0.64)。

表3 8种雌激素类药物标准曲线及相关系数
2.4 方法的灵敏度、回收率和精密度 对猪和鸡的空白饲料样品中添加适量标准溶液各进行3次添加回收试验,每次每个浓度点进行5个样品的重复。从表4中可见,2种配合饲料中8种雌激素在1~50 μg/kg添加浓度范围内的平均添加回收率为97.5%~106%,批间RSD在4.3%~7.9%。同时以3倍信噪比(S/N)对应的样品中雌激素浓度作为检出限(LOD),10倍信噪比(S/N)对应的样品中雌激素浓度作为定量限(LOQ),得到各雌激素的检出限和定量限。由表4可见,本方法的检测限和定量限范围分别为0.15~0.30 μg/kg和0.50~1.0 μg/kg。猪配合饲料空白样品和添加样品详见图1,其中b6中17α-雌二醇出峰时间在后,17β-雌二醇出峰时间在前。
2.5 方法应用 利用该方法测定了20个猪配合饲料和15个鸡配合饲料样品,8种雌激素类药物均未检出。
3 讨 论
复杂的前处理步骤是饲料和动物性食品中建立高灵敏度、高分析通量分析方法的关键制约因子。林小莉等[3]建立的饲料中雌激素类药物分析方法前处理中包括了有机溶剂提取-氢氧化钠溶液液液萃取净化-固相萃取净化和衍生化,整个前处理不仅繁琐耗时,单个样品前处理时间在3 h以上;而且会导致方法重复性较差,方法的批间RSD为6.7%~22.7%。本研究所建立的前处理步骤相对非常简单,仅包括乙腈提取-碳酸钠溶液液液萃取净化2步,其中液液萃取净化步骤所需时间只有10 min左右,不仅远快于林小莉等建[3]立的净化方法,也比目前很多LC-MS/MS方法中建立的常规固相萃取方法[9,13-14,16-17](通常需要1 h左右)要节约80%以上的时间,基本和目前QuEChERS净化步骤[4,15]所需时间相当。
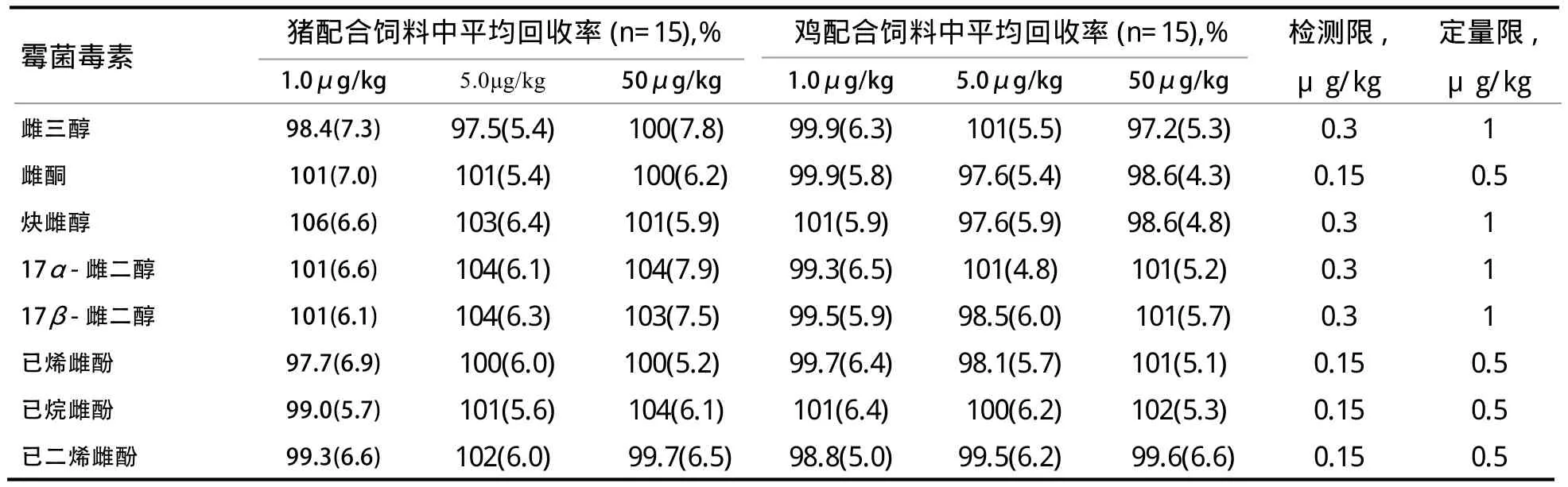
表4 8种雌激素类药物在猪、鸡配合饲料中的灵敏度、回收率和精密度

图 1 猪配合饲料空白样品(a1-7)和添加样品(b1-7, 1.0μg/kg)MRM色谱图
液质联用中采用同位素内标物定量不仅能最大限度减少样品损失造成的分析误差,而且能够克服基质效应造成的定量误差。本方法对于8种雌激类药物均采用一一对应的8种同位素内标物进行定量,不仅明显好于未采用内标法定量的LCMS/MS方法,如张艳等[13]建立的液质方法的平均回收率为75.3 %~120.0%,相对标准偏差(RSD)为3.4%~19.6%;而且也明显好于虽采用内标法定量,但仅用1个内标物来对多种雌激素定量液质联用分析方法,如王和兴等[18]采用17-β-雌二醇-D3对9种雌激素类药物定量建立的LC-QTOF-MS方法的平均回收率为61%~137%,RSD 为1.0%~22.6%(其中17-β-雌二醇的平均回收率为102%~110%,RSD为1.2%~10%)。
4 小 结
通过优化前处理和仪器条件,本研究建立了准确度好、灵敏度高和分析速度快的饲料中8种雌激素类药物LC-MS/MS同步分析方法;同时通过样品添加和实际样品分析,该分析方法适用于饲料中雌激素类药物的定性定量分析。
[1] 孟萌, 夏雅娟, 李建云. 雌激素受体与癌症的研究进展[J].内蒙古医学杂志, 2008, 40(8): 835-837.
[2] 卢琰. 雌激素受体在卵巢癌研究中的进展探析[J]. 世界最新医学信息文摘, 2015, 15(59): 31-32.
[3] 林小莉, 李宁, 霍峰, 等. 气相色谱-质谱法同时测定饲料中6 种雌激素类药物[J]. 分析测试学报, 2016, 35(3):322-326.
[4] 马丽莎 , 戴晓欣 , 谢文平 , 等 . QuEChERS /GC-MS 法同时测定鱼、虾中的雌二醇与己烯雌酚残留[J]. 分析测试学报, 2015, 34(1): 62-66.
[5] 孙汉文, 李挥, 康占省, 等. 气相色谱-质谱法同时检测婴幼儿配方奶粉中的6种雌激素残留[J]. 河北大学学报(自然科学版), 2011, 31(6): 607-611.
[6] Daeseleire E, Vandeputte R, Van Peteghem C. Validation of multi-residue methods for the detection of anabolic steroids by GC-MS in muscle tissues and urine samples from cattle[J]. The Analyst, 1998, 123(12): 2595-2598.
[7] Quintana J B, Carpinteiro J, Rodriguez I, et al. Determination of natural and synthetic estrogens in water by gas chromatography with mass spectrometric detection[J]. J Chromatogr A, 2004, 1024(1-2): 177-185.
[8] 尹江伟, 刘红河, 刘祖强, 等. 高效液相色谱-串联质谱法测定肉类食品和饲料中雌二醇和雌三醇[J]. 现代预防医学, 2010, 37(10): 1928-1933.
[9] 尤亮亮, 魏巍, 刘海燕, 等. 固相萃取-高效液相色谱串联质谱测定牛奶中9种性激素残留[J]. 中国测试, 2016,42(5): 46-49.
[10] Dong Z, Wang C, Zhang J, et al. A UHPLC-MS/MS method for profiling multifunctional steroids in human hair[J]. Anal Bioanal Chem, 2017, 409(20): 4751-4769.
[11] Wang C, Wu C, Zhang L, et al. Ultraperformance liquid chromatography-tandem mass spectrometry method for profiling ketolic and phenolic sex steroids using an automated injection program combined with diverter valve switch and step analysis[J]. Anal Chem, 2016, 88(16):7878-7884.
[12] Weisser J J, Hansen C H, Poulsen R, et al. Two simple cleanup methods combined with LC-MS/MS for quantification of steroid hormones in in vivo and in vitro assays[J]. Anal Bioanal Chem, 2016, 408(18):4883-4895.
[13] 张艳, 陈剑刚, 冯翠霞. 液相色谱-串联质谱法同时测定牛奶中7 种雌激素类药物残留[J]. 实用预防医学, 2013,20(6): 740-743.
[14] 徐英江, 田秀慧, 张秀珍, 等. 超高效液相色谱串联质谱法对水产品中8种雌激素的测定[J]. 分析测试学报,2010, 29(2): 152-156.
[15] Socas-Rodríguez B, Lanková D, Urbancová K, et al.Multiclass analytical method for the determination of natural/synthetic steroid hormones, phytoestrogens, and mycoestrogens in milk and yogurt[J]. Anal Bioanal Chem,2017, 409(18): 4467-4477.
[16] Capriotti A L, Cavaliere C, Foglia P, et al. Ultra-highperformance liquid chromatography-tandem mass spectrometry for the analysis of free and conjugated natural estrogens in cow milk without deconjugation[J]. Anal Bioanal Chem, 2015, 407(6): 1705-1719.
[17] Cavaliere C, Capriotti A L, Foglia P, et al. Natural estrogens in dairy products: determination of free and conjugated forms by ultra high performance liquid chromatography with tandem mass spectrometry[J]. J Sep Sci, 2015, 38(20):3599-3606.
[18] 王和兴, 周颖, 姜庆五. 超高效液相色谱-四极杆飞行时间串联质谱法同时分析奶粉中9 种雌激素[J]. 分析化学,2011, 39(9): 1323-1328.
Simultaneous Determination of Estrogen Drugs in Feeds by Liquid Chromatography-Tandem Mass Spectrometry with Isotope Dilution
XUE Yi1, ZHANG Yue1, LV Yan2, WU Yin-Liang2*
(1. The Center for Animal Disease Control and Prevention, Beijing 100125, China;2. The Ningbo Academy of Agricultural Sciences, Zhejiang Ningbo 315040, China)
A method was developed for determining eight estrogen drugs in feeds using liquid chromatography tandem mass spectrometry with isotope dilution. Samples were extracted with acetonitrile and centrifuged. The extract was dried under nitrogen and the residue was redissolved with ethyl acetate. Then, the samples were cleaned up by sodium carbonate solution with a liquid-liquid extraction method. After clean-up, the samples were analyzed by LC-MS/MS on an Acquity HSS T3 column with a mixture of water and acetonitrile as mobile phase under gradient elution conditions. The analytes were determined by negative electrospray ionization with multiple reaction monitoring (MRM). Internal standard method was used to quantify the analytes. There is a good linear correlation between the peak areas and concentration of eight estrogen drugs in the range of 1.0 μg/L to 200 μg/L. The eight estrogen drugs were measured in fortified feeds at 1.0~50 μg/kg levels. Average recoveries were in the range of 97.5%~106%. The limit of detection (LOD) and limit of quantification (LOQ) was 0.15~0.30 μg/kg and 0.50~1.0 μg/kg, respectively. The method is demonstrated to be suitable for the determination of the eight estrogen drugs in feeds.
Estrogen drugs; Feed; Isotope dilution; Liquid chromatography tandem mass spectrometry
O657.63
A
10.19556/j.0258-7033.2017-10-101
2017-07-10;
2017-08-18
宁波市农业科技攻关项目(2013C11003、2015C50067)作者简介:薛毅(1972-),男,硕士,兽医师,研究方向为兽药安全性评估,E-mail:xingmuyikang@163.com
* 通讯作者:吴银良(1975-),男,博士,教授级高工,研究方向为农产品中农兽药残留分析研究,E-mail:wupaddyfield@sina.com
