活性炭促进玉米幼胚离体培养幼苗生长与发育的转录组分析
2017-09-25王金萍孙果忠王海波河北农业大学农学院河北保定0700河北省农林科学院遗传生理研究所河北省植物转基因中心河北石家庄05005
王金萍孙果忠王海波河北农业大学农学院, 河北保定 0700;河北省农林科学院遗传生理研究所/河北省植物转基因中心, 河北石家庄 05005
活性炭促进玉米幼胚离体培养幼苗生长与发育的转录组分析
王金萍1,2孙果忠2,*王海波2,*
1河北农业大学农学院, 河北保定 071001;2河北省农林科学院遗传生理研究所/河北省植物转基因中心, 河北石家庄 050051
以玉米自交系昌7-2为试材, 比较了14 d龄幼胚在MS和MSA (MS加活性炭)培养基上离体培养9 d的幼苗生长情况, 并利用转录组测序技术分析了2种培养基上的玉米幼苗地上部和地下部的基因表达差异。结果表明, 活性炭可以显著促进玉米幼苗的生长与发育。活性炭主要影响地下部基因表达。加入活性炭后, 幼苗地下部表达上调的基因有1612个, 下调的基因有530个; 幼苗地上部表达上调的基因有69个, 下调的基因有78个。GO功能显著性分析表明, 地下部差异表达基因(DEGs)主要涉及DNA包装、DNA包装复合体和水解酶活性, 地上部DEGs主要涉及脂类代谢、胞外区和过氧化物酶活性。KEGG富集分析表明, 地下部 DEGs主要涉及能量、碳水化合物、脂类和氨基酸代谢, 以及细胞周期和植物激素转导途径; 地上部 DEGs主要涉及泛醌和其他萜-醌类物质合成途径。活性炭促进细胞周期途径中的关键基因(CYC、CDH1、MCM3、PCNA2和BUBR1)、生长素信号传导基因(Aux/IAA)以及细胞色素基因(CYP450)显著上调表达。利用实时荧光定量PCR (qRT-PCR)验证了10个DEGs的表达模式与转录组测序结果一致, 证实了转录组数据与分析的可靠性。
玉米; 幼胚; 活性炭; 转录组
利用幼胚离体培养直接发育成苗可以节省种子发育成熟的耗时, 显著缩短植物发育周期, 有助于加快育种进程[1], 建立以幼胚培养为关键步骤的植物快速加代技术对促进植物育种和遗传学研究具有重要意义[2-4]。在适宜条件下, 种子膨胀吸水, 预存的代谢系统重新活化, 细胞开始伸长、分裂至胚根突破种皮完成萌发, 随后进入幼苗形态建成阶段[5]。种子萌发伴随着一系列有序的生理和形态发生过程,包括呼吸作用加强、能量和物质代谢的激活、DNA修复和蛋白质的合成等[6]。种子中贮藏mRNA的翻译对种子萌发非常关键, 抑制翻译过程, 则萌发停止[7-9]。种子萌发过程还受植物激素的调控, 如赤霉素(GA)和脱落酸(ABA)。ABA主要在种子萌发早期阶段起抑制作用, 萌发条件适宜时, ABA逐渐减少,萌发顺利进行[10-12]。GA促进种子萌发主要在胚根突破种皮阶段, 使β-糖苷酶、膨胀素基因的表达上调[13]。
活性炭(active carbon, AC)是植物微繁殖、组织培养、体细胞胚发生和胚胎培养等过程中常用的添加物[14-16]。一般认为 AC通过其吸附功能和创造培养基的暗环境, 影响离体培养的效果。AC吸附功能很强, 可以吸附培养基中的有害物质, 如琼脂中的杂质、蔗糖高压消毒过程中产生的 5-羟甲基糠醛(5-hydroxymethy-lfurfural)[14]、离体培养过程中产生的有毒代谢物、酚醛类分泌物[17-19]、生长调节物质[20-21]、金属离子[22]等。强光对生长素(如IAA和IBA)具有破坏作用, AC可通过创造培养基暗环境, 减缓培养基中IAA和IBA的降解[23]。
转录组测序是揭示某一物种特定组织或器官在某一状态下的几乎所有转录本序列信息的重要技术手段[24-26]。为揭示AC影响幼胚成苗的分子机制, 本研究利用转录组测序技术分析了在 MS[27]和 MSA (MS加 AC)培养基上离体培养的玉米幼胚苗地上部和地下部的基因表达情况, 并对部分表达差异显著的基因进行了实时荧光定量RT-PCR的验证, 旨在揭示AC促进玉米幼胚发育成苗的基因表达机制, 为进一步改进玉米幼胚离体萌苗技术提供依据。
1 材料与方法
1.1 植物材料及处理
将玉米自交系昌 7-2种子播于温室, 温度控制在23~30℃, 自然光照。吐丝前套袋, 人工授粉14 d后, 取幼穗在无菌条件下剥取幼胚, 盾片朝下接种于MS和MSA培养基上。
MSA培养基为添加AC 6 g L–1的MS培养基。MS和MSA两种培养基均附加0.01 mg L–1NAA、0.04 mg L–16-BA、20 g L–1蔗糖和6 g L–1琼脂。培养基分装于100 mL三角瓶中, 每瓶约30 mL。每瓶接种6个幼胚, 每种培养基接种5瓶, 2个重复。接种后的幼胚培养于24℃、16 h光照/8 h黑暗的培养箱中。生长9 d后, 从每瓶发育较为一致的幼苗中取2株, 调查幼苗根长(最长根的长度)、根数、株高和单株鲜重, 对每种培养基处理调查10株, 2个重复。利用SPSS软件对数据进行两个独立样本的 t检验,分析性状的差异显著性。从每种培养基上选取生长一致的4株幼苗按地上部(茎和叶)和地下部(残存盾片和根)取样, 共4个样品, 即MS-shoots、MS-roots、MSA-shoots和MSA-roots, 3个生物学重复。样品经液氮速冻后, -80℃保存, 用于RNA提取。
1.2 转录组测序
委托上海派森诺公司完成RNA提取、文库构建和转录组测序。利用TRIZOL试剂提取样品总RNA,通过rRNA去除试剂盒纯化得到mRNA, 构建链特异性文库。采用Illumina NextSeq 500平台进行双末端测序, 得到原始序列(raw reads)。
1.3 基因表达数据分析
经过去除接头和低质量reads的数据过滤, 得到Q > 20的高质量clean reads序列用于后续分析。参考基因组数据来源于 Ensembl数据库(http://www. ensembl.org/)[28]。通过Bowtie2软件建立参考基因组索引, 用Tophat2将clean reads比对到参考基因组上[29-30]。用HTSeq统计比对到每个基因上的Read Count值,作为基因的原始表达量[31]。用RPKM (Reads Per Kilo bases per Million reads)对表达量进行标准化, 以RPKM值 > 1为基因表达标准。用DESeq对基因表达进行差异分析[32-33], 以表达倍数差异Fold change值 > 2、显著性P-value < 0.05和FDR < 0.05为差异基因筛选条件。用 GO功能富集分析对差异基因(differentially expressed genes, DEGs)进行功能注释,显示DEGs显著富集的功能分类[33]。用KEGG进行生物通路分析确定 DEGs主要参与的代谢途径和信号通路[35-36]。
1.4 荧光定量RT-PCR验证
通过分析转录组数据, 选取 2种培养基上生长的幼胚苗地下部的10个DEGs, 利用Primer 5.0软件设计候选基因引物(表1)。以玉米甘油醛-3-磷酸脱氢酶(GAPDH)的编码基因为内参, 进行荧光定量RT-PCR验证。以RNA为模板, 用FastQuant cDNA第一链合成试剂盒(天根公司)合成 cDNA。用SuperReal荧光定量预混试剂(天根公司)配制反应液,用7500 Real-time PCR System进行荧光定量PCR,反应程序为95℃ 30 s; 95℃ 5 s, 60℃ 32 s, 40个循环。以 2–ΔΔCT方法计算基因的相对表达量。对每个基因的定量设置3个生物学重复。以MS-roots样品中基因表达量为1, 统计MSA-roots中各基因的相对表达量, 用单样本t检验分析MSA-roots样本中10个DEGs表达量是否达到显著水平(P < 0.05)。分析RNA-Seq和qRT-PCR两平台中10个DEGs相对表达量的相关性。
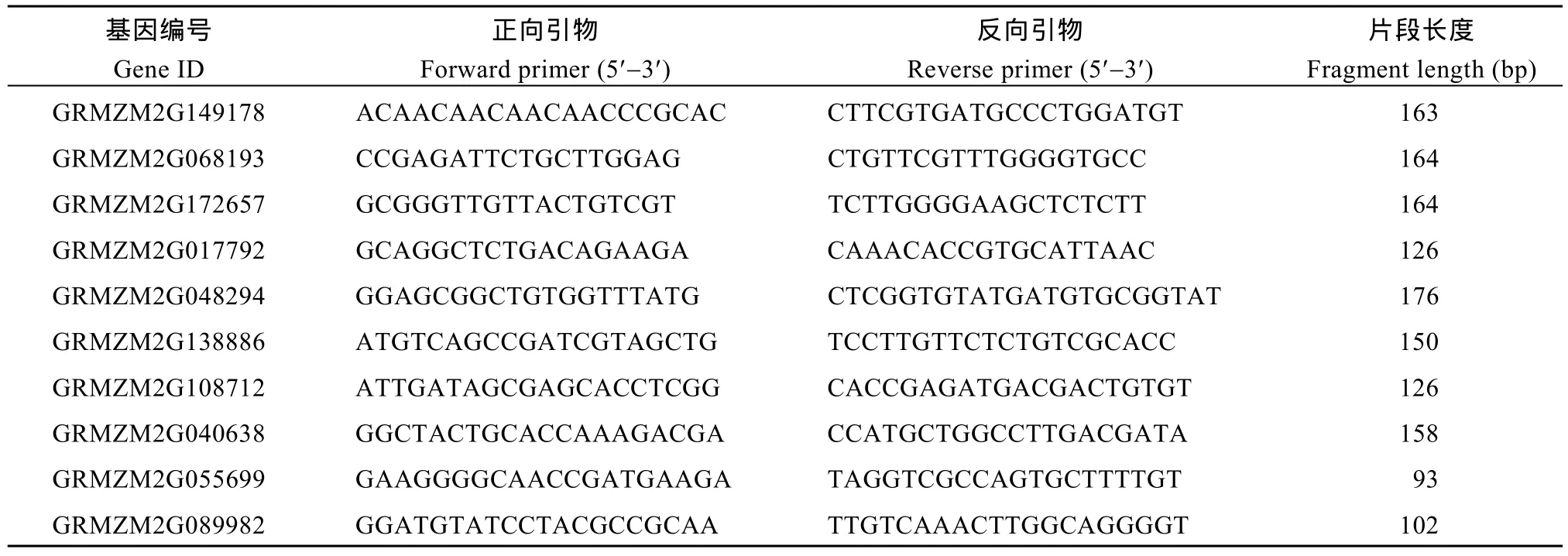
表1 用于qRT-PCR分析的10个基因及其引物Table 1 Primers used for qRT-PCR analysis of ten maize DEGs
2 结果与分析
2.1 AC显著促进玉米幼胚苗的生长发育
胚龄14 d的玉米幼胚在MS和MSA培养基上培养9 d后, 幼苗长势差异明显(图1)。MSA培养基上的幼苗根长、根数、株高和鲜重均显著高于 MS培养基上的幼苗(P < 0.05)(表 2)。在培养基中添加AC可以显著促进玉米幼胚离体培养成苗。
2.2 转录组数据分析
MS-roots、MS-shoots、MSA-roots和MSA-shoots分别有41 572 394、47 070 088、44 177 188和44 369 556个clean reads。共有130 209 524个clean reads比对到参考基因组上, 115 713 895个clean reads比对到基因区域, 其中96.96%比对到外显子区域(表3)。
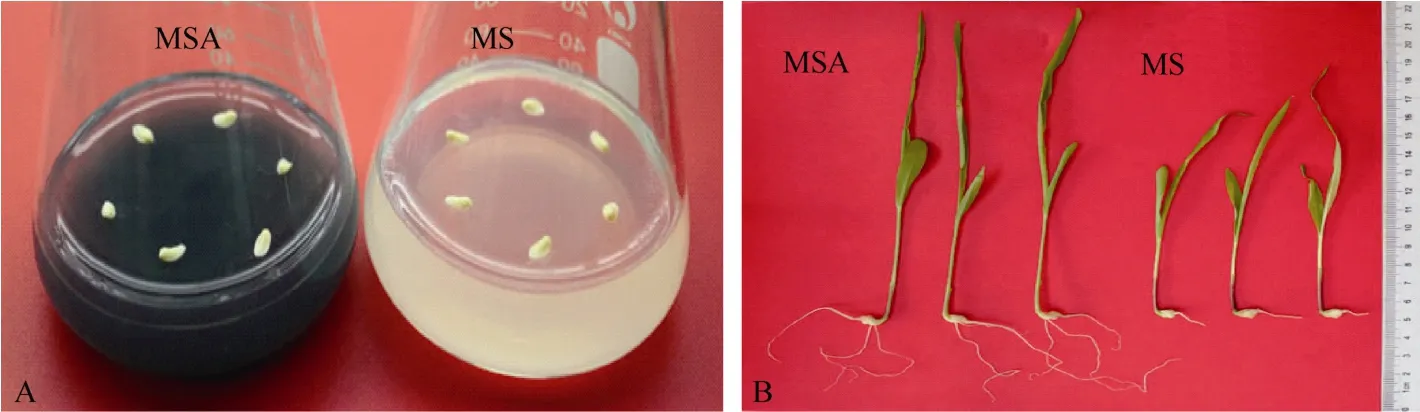
图1 活性炭促进玉米幼胚离体培养成苗Fig. 1 Active carbon accelerated the seedlings producing from immature embryos cultured in vitro
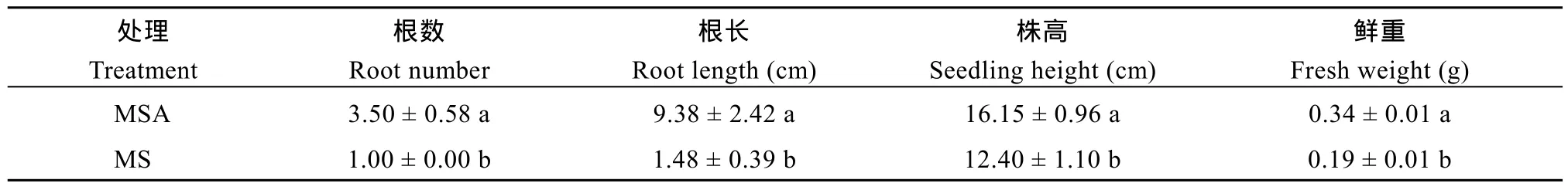
表2 MS和MSA培养基上玉米幼胚苗性状的比较Table 2 Comparison of the traits of maize seedlings developing from immature embryos cultured on MS or MSA media
2.3 AC影响玉米幼胚苗地上部和地下部的基因表达
对MS和MSA培养基上玉米幼胚苗的基因表达进行分析, 共发现2196个DEGs。其中, 地下部2142个, 地上部147个; 有93个基因在地上部和地下部中同时存在(图2-A)。在2142个地下部DEGs中, 有1612个基因受AC上调表达, 530个基因受AC下调表达。在147个地上部DEGs中, 有69个基因受AC上调表达, 78个基因受AC下调表达(图2-B)。这些结果表明, AC主要影响玉米胚苗地下部基因的表达。
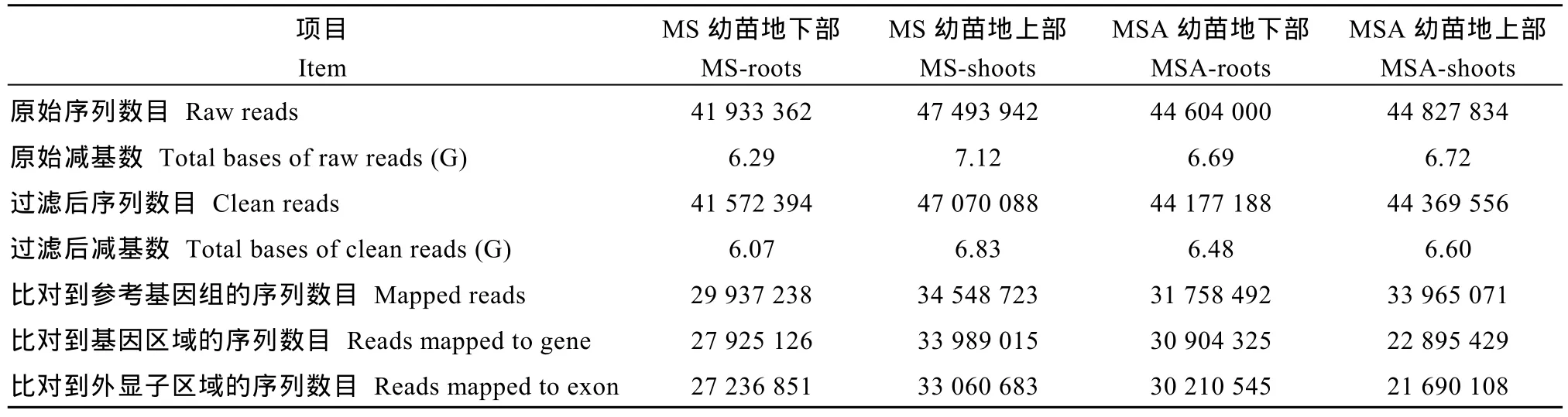
表3 玉米幼胚苗的转录组数据分析Table 3 Analysis on transcriptome data of maize immature embryo seedlings
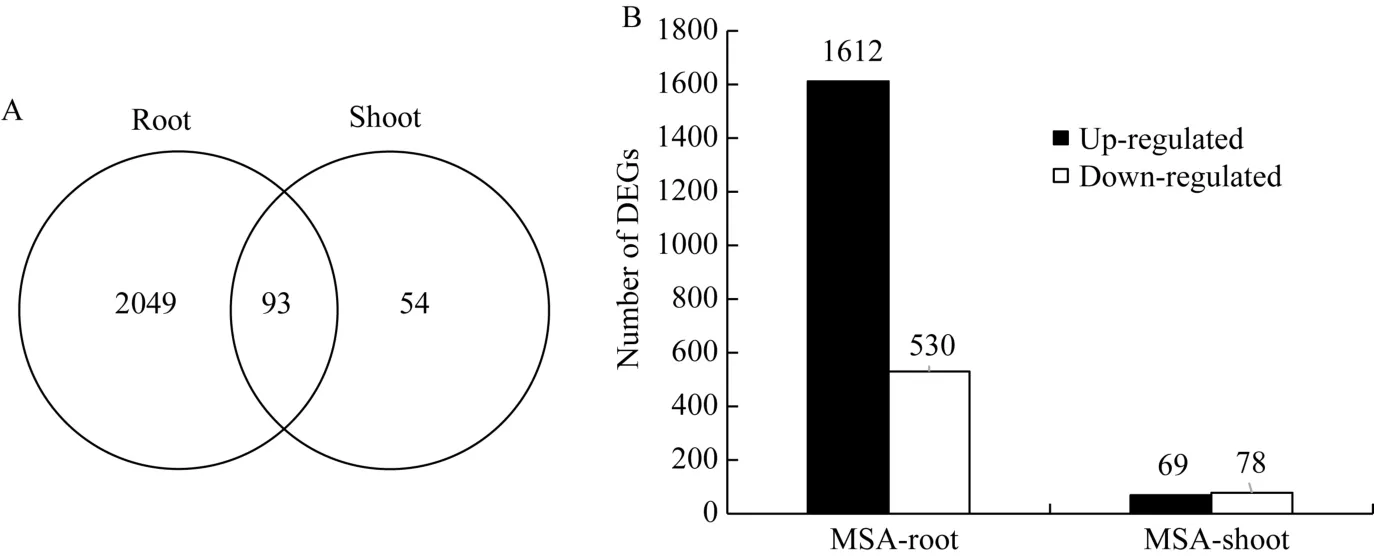
图2 活性炭诱导的差异表达基因分布情况Fig. 2 Distribution of DEGs induced by active carbon
2.4 AC诱导的DEGs的GO富集分析
GO功能富集分析表明, 地下部的2142个DEGs中有 1630个基因获得 GO注释, 涉及生命进程(biological process, BP)、细胞组分(cellular component, CC)和分子功能(molecular function, MF)三大类生物功能; 进一步可细分成 131个显著富集小类(terms)(P < 0.05)。其中, DNA组装(P = 4.36×10-19)、DNA组装复合体(P = 7.75×10-18)和糖基水解酶活性(P = 9.02×10-13)分别是BP、CC和MF中最显著富集的功能类别。地上部142个DEGs中有108个被注释, 分别有16个BP、5个CC和15个MF Terms显著富集(P < 0.05), 最显著富集的类别涉及脂类代谢过程(P = 4.25×10-6)、胞外区(P = 3.00×10-7)和过氧化物酶活性(P = 4.40×10-4)。地下部和地上部中DEGs涉及的20个最显著富集的功能见图3。
2.5 AC诱导的DEGs的KEGG富集分析
KEGG富集分析表明, 地下部DEGs注释到108个代谢通路。其中, 56个涉及能量、碳水化合物、氨基酸、脂类和次生代谢物等代谢途径; 11个涉及细胞周期、p53信号通路、肌动蛋白细胞骨架调节等细胞进程途径; 13个涉及植物激素信号转导、MAPK信号通路和 AMPK信号通路等环境信息进程途径; 8个涉及DNA复制、碱基切除修复和核糖体等遗传信息进程途径; 20个涉及黄体酮调节卵母细胞成熟、植物-病原菌互作、趋化因子信号通路等有机系统途径。20个最显著富集的代谢通路主要是光合作用-天线蛋白、苯丙烷类物质生物合成和细胞周期途径。地上部DEGs涉及10个代谢通路, 最显著富集途径为泛醌和其他萜-醌类物质合成(图4)。
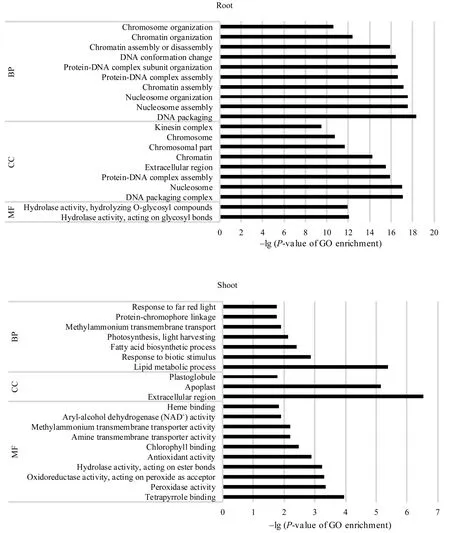
图3 活性炭诱导的玉米胚苗地上部和地下部DEGs中各涉及的20个最显著富集GO分类Fig. 3 Twenty most significantly enriched Gene Ontology (GO) terms of the DEGs induced by active carbon in roots and shoots of maize seedlings
对地下部富集的细胞周期途径、植物激素转导途径和外源物质代谢途径中的DEGs进一步分析。细胞周期途径涉及 7个 DEGs上调表达, 分别为细胞周期蛋白 A2、细胞周期蛋白 B2、细胞周期超家族蛋白、细胞分裂周期蛋白 CDH1、增殖细胞核抗原PCNA、微小染色体维持缺陷蛋白MCM3和有丝分裂检纺锤体监测点蛋白 BUBR1的编码基因。在植物激素转导途径中, 生长素信号转导中的 2个Aux/IAA编码基因上调表达, 水杨酸信号转导中的具有bZIP结构的TGA转录因子基因下调表达。在外源物质代谢途径中, 细胞色素 P450、乙醛脱氢酶3B1、谷胱氨肽转移酶7和谷胱氨肽转移酶27的编码基因表达上调, 乙醇脱氢酶2基因表达下调(表4)。
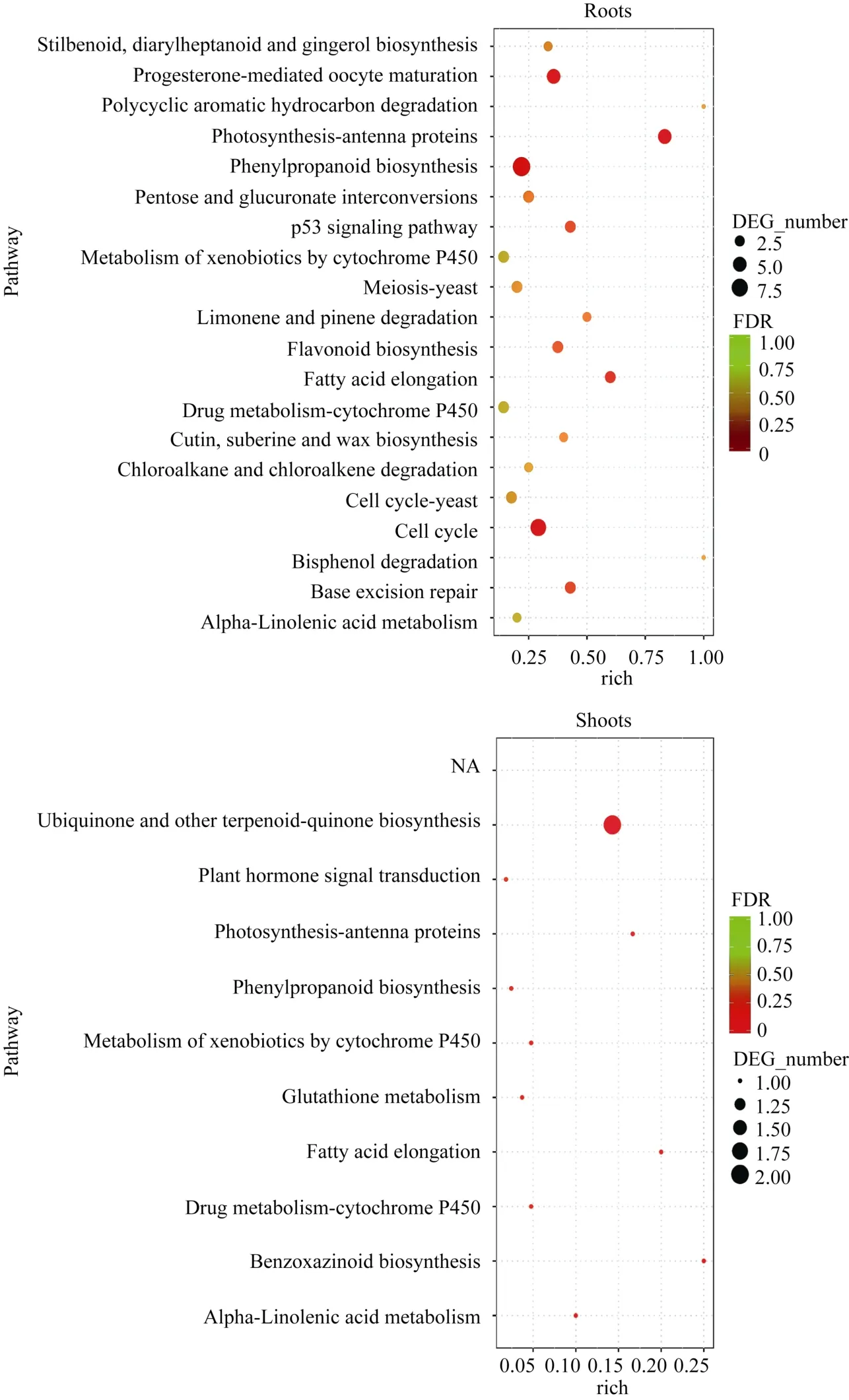
图4 地下部和地上部样品中DEGs富集的KEGG途径Fig. 4 Enriched KEGG pathways of DEGs in the roots and shoots samples
2.6 DEGs的实时荧光定量RT-PCR验证
对 2种培养基上生长的幼胚苗地下部中 10个DEGs进行了荧光定量 RT-PCR验证。以 MS-roots样品中基因表达量为1, 统计MSA-roots中各基因的相对表达量。结果表明, qRT-PCR中的基因表达变化与 RNA-seq结果一致(图 5)。经单样本 t检验, MSA-roots样品中10个DEGs表达量与MS样品中表达量均差异显著。将RNA-seq和qRT-PCR中基因差异表达量进行 log2转化后进行相关性分析, 皮尔森相关系数为0.922, 在0.01水平上显著相关(图6),说明RNA-seq的结果可靠。
经 qRT-PCR验证, 组蛋白 H4 (GRMZM2
G149178)、细胞周期蛋白 CYCB2 (GRMZM2G 138886)、细胞周期蛋白激酶 CDK (GRMZM2G 068193)、增殖细胞核抗原 PCNA2 (GRMZM2G 108712)、木质素过氧化物酶 POX72 (GRMZM2G 089982)、GRAS转录因子 GRAS19 (GRMZM2G1 72657)和富亮氨酸重复类受体蛋白激酶 LRR-RLK (GRMZM2G048294)受 AC影响在地下部中上调表达。MAP激酶(GRMZM2G017792)、木质素过氧化物酶 POX2 (GRMZM2G089982)和 β-糖苷酶(GRM ZM2G055699)受AC影响在地下部中下调表达。

表4 细胞周期、植物激素转导和外源物质降解代谢途径中的DEGsTable 4 DEGs functioning in cell cycle, plant hormone signal transduction, xenobiotics bio-degradation and metabolism pathways
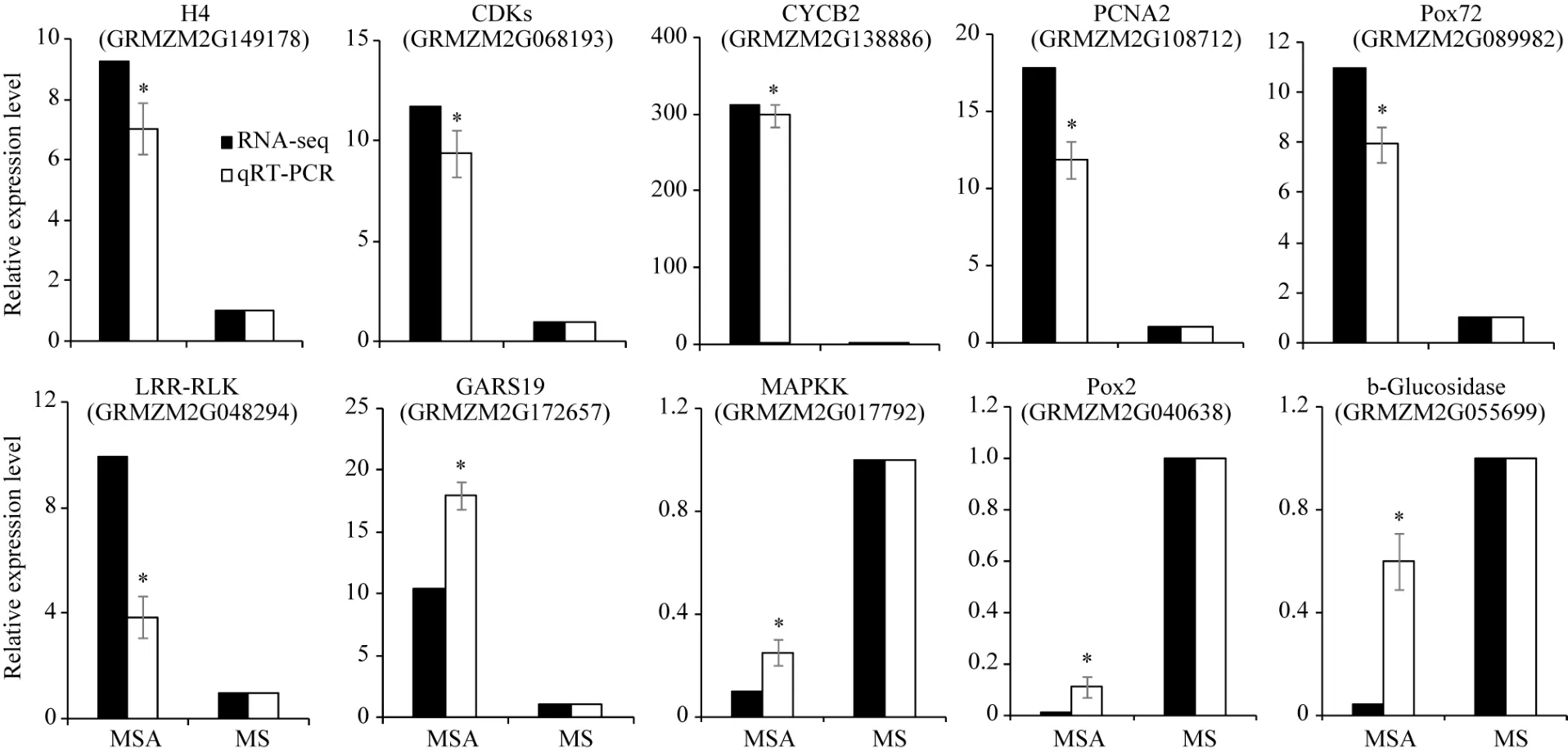
图5 受活性炭诱导的10个DEGs的qRT-PCR检测Fig. 5 qRT-PCR verification of ten DEGs induced by active carbon
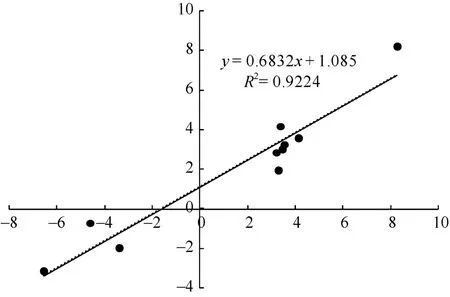
图6 10 DEGs的RNA-seq与qRT-PCR结果相关性分析Fig. 6 Correlation analysis of the expression changes of ten DEGs as revealed by RNA-seq and qRT-PCR, respectively
3 讨论
本研究利用Illumina 500测序技术对AC促进玉米幼胚发育成苗的基因表达情况进行了转录组分析。测序数据质量检测表明, 高质量数据(Q20)在97.34%以上, 可以保证后续分析结果的准确性。选取的10个DEGs的qRT-PCR结果与转录组分析一致, 表明转录组测序结果可信度高。
AC促进玉米胚苗的生长发育可能与控制细胞分裂的基因调控表达上调有密切关系。本研究中, AC除了影响地下部细胞周期途径中的CYC、CDH、PCNA、MCM、BUB基因上调表达外, 还影响其他与细胞增殖相关基因显著上调表达, 如H4、CDK、GRAS等。前人研究表明, 组蛋白是染色体的组成部分, 其翻译后修饰可引起染色质结构重塑, 在基因表达调控中起重要作用[37-38]。CYC和 CDK通过特异性结合调控细胞周期, 控制细胞增殖, 是细胞周期调控的主要因子[39]。PCNA是DNA多聚酶的一种辅助蛋白质, 在DNA复制、DNA修复和细胞周期中发挥重要作用, 是细胞增殖活性的重要指标[40-42]。 MCM是与DNA复制和细胞增殖密切相关的蛋白家族, 在真核细胞的DNA复制中, 形成复制前复合物,具有DNA解链酶活性, 启动DNA复制, 是调控细胞由G1期进入S期的关键蛋白, 是反映细胞增殖活性的特异性指标[43-44]。GRAS转录因子是植物特有的转录因子, 在植物生长发育、信号转导、解毒、生物和非生物胁迫相关应答过程中起作用[45-46]。上述结果表明, 在培养基中添加 AC可以调控细胞增殖相关的基因表达, 从而促进玉米幼胚苗的生长与发育。
激素对植物发育具有重要的调节作用。本研究中AC促进幼胚苗根部生长素(Auxin)信号转导途径中Aux/IAA基因上调表达。Auxin影响胚发生、根发育、叶片伸展和果实成熟等许多生长发育过程[47-48]。Aux/IAA蛋白在Auxin信号转导途径中起主要的调控作用, Auxin浓度低时, Aux/IAA蛋白与生长素反应因子ARF结合, 抑制下游基因的表达, Auxin浓度高时, Aux/IAA蛋白泛素化降解, ARF激活下游基因表达[49]。本研究中AC促进水杨酸(salicylic acid, SA)信号转导途径中 TGA转录因子基因下调表达。SA作为植物内源信号分子, 主要参与应对生物和非生物胁迫过程, 提高植物抵抗能力[50]。TGA是SA信号转导途径中的重要转录因子, 与病程相关蛋白基因非表达子 1 (nonexpressor of pathogenesis-related protein gene 1, NPR1)相互作用, 激活病程相关蛋白1 (pathogenesis-related protein 1, PR1)表达, 最终诱导系统获得性抗性(systemic acquired resistance, SAR)的产生[51-52]。AC 可以促进胚苗地下部中CYP450编码基因的上调表达。CYP450是一种末端加氧酶, 作为植物中最大的酶家族广泛参与植物体内多种物质合成和降解过程, 在催化外源物质降解过程中具有重要作用[53]。
4 结论
发现 AC能显著促进玉米幼胚离体培养幼苗的生长和发育, 并从转录组水平上揭示了 AC促进玉米幼胚离体成苗的基因表达情况。AC可引起玉米幼胚苗中2196个基因的差异表达, 包括地下部的2142个基因和地上部的147个基因。地下部中DEGs主要涉及光合作用-天线蛋白、细胞循环、植物激素信号转导、外源物质代谢等108条KEGG途径; 地上部DEGs涉及10条KEGG途径, 最显著富集的为泛醌和其他萜-醌类物质合成途径。上述结果对揭示玉米幼胚萌发的分子机理和进一步改进玉米幼胚离体培养条件提供了依据。
[1] 王海波, 王彦霞, 赵和. 如何加快作物遗传改良的速度. 河北农业科学, 2003, (7): 50–56 Wang H B, Wang Y X, Zhao H. How to accelerate the process of plant genetic modification. J Hebei Agric Sci, 2003, 7: 50–56 (in Chinese with English abstract)
[2] Forster B P, Till B J, Ghanim A M A, Huynh H O A, Burstmayr H, Caligari P D S. Accelerated plant breeding. CAB Rev, 2014, 9: 1–16
[3] Yao Y, Zhang P, Wang H B, Lu Z Y, Liu C J, Liu H, Yan G J. How to advance up to seven generations of canola (Brassica napus L.) per annum for the production of pure line populations. Euphytica, 2016, 209: 113–119
[4] Liu H, Zwer P, Wang H B, Liu C J, Lu Z Y, Wang Y X, Yan G J. A fast generation cycling system for oat and triticale breeding. Plant Breed, 2016, 135: 574–579
[5] Bewley J D. Seed germination and dormancy. Plant Cell, 1997, 9: 1055–1066
[6] Bove J, Jullien M, Grappin P. Functional genomics in the study of seed germination. Genome Biol, 2002, 3: 1002.1–1002.5
[7] Galland M, Huguet R, Arc E, Cueff G, Job D, Rajjou L. Dynamic proteomics emphasizes the importance of selective mRNA translation and protein turnover during Arabidopsis seed germination. Mol Cell Proteomics, 2014, 13: 252–268
[8] Rajjou L, Gallardo K, Debeaujon I, Vandekerckhove J, Job C, Job D. The effect of α-amanitin on the Arabidopsis seed proteome highlights the distinct roles of stored and neosynthesized mRNAs during germination. Plant Physiol, 2004, 134: 1598–1613
[9] Abraham Z, Fernández R I, Martinez M, Diaz I, Carbonero P, Vicente-Carbajosa J. A developmental switch of gene expression in the barley seed mediated by HvVP1 (Viviparous 1) and HvGAMYB interactions. Plant Physiol, 2016, 170: 2146–2158
[10] Miransari M, Smith D L. Plant hormones and seed germination. Environ Exp Bot, 2014, 99: 110–121
[11] Umezawa T, Nakashima K, Miyakawa T, Kuromori T, Tanokura M, Shinozaki K, Yamaguchi-Shinozaki K. Molecular basis of the core regulatory network in ABA responses: sensing, signaling and transport. Plant Cell Physiol, 2010, 51: 1821–1839
[12] Weitbrecht K, Müller K, Leubner-Metzger G. First off the mark: early seed germination. J Exp Bot, 2011, 62: 3289–3309
[13] Sun T. Gibberellin-GID1-DELLA: a pivotal regulatory module for plant growth and development. Plant Physiol, 2010, 154: 567–570
[14] Pan M J, van Staden J. The use of charcoal in in vitro culture: a review. Plant Growth Regul, 1998, 26: 155–163
[15] Thomas T D. The role of activated charcoal in plant tissue culture. Biotechnol Adv, 2008, 26: 618–631
[16] Mittal P, Devi R, Gosal S S. Effect of genotypes and activated charcoal on high frequency in vitro plant regeneration in sugarcane. Ind J Biotechnol, 2016, 15: 261–265
[17] Fridborg G, Pedersén M, Landström L E, Eriksson T. The effect of activated charcoal on tissue cultures: adsorption of metabolites inhibiting morphogenesis. Physiol Plant, 1978, 43: 104–106
[18] Manchanda P, Gosal S S. Effect of activated charcoal, carbon sources and gelling agents on direct somatic embryogenesis and regeneration in sugarcane via leaf roll segments. Sugar Tech, 2012, 14: 168–173
[19] Nguyen T V, Thu T T, Claeys M, Angenon G. Agrobacterium-mediated transformation of sorghum (Sorghum bicolor (L.) Moench) using an improved in vitro regeneration system. Plant Cell Tissue Organ Cult, 2007, 91: 155–164
[20] Ebert A, Taylor F, Blake J. Changes of 6-benzylaminopurine and 2,4-dichlorophenoxyacetic acid concentrations in plant tissue culture media in the presence of activated charcoal. Plant Cell Tissue Organ Cult, 1993, 33: 157–162
[21] Ebert A, Taylor H F. Assessment of the changes of 2, 4-dichlorophenoxyacetic acid concentrations in plant tissue culture media in the presence of activated charcoal. Plant Cell Tissue Organ Cult, 1990, 20: 165–172
[22] Halhouli K A, Darwish N A, Al-Dhoon N M. Effects of pH and inorganic salts on the adsorption of phenol from aqueous systems on activated decolorizing charcoal. Separat Sci Technol, 1995, 30: 3313–3324
[23] Nissen S J, Sutter E G. Stability of IAA and IBA in nutrient medium to several tissue culture procedures. Hortscience, 1990, 25: 800–802
[24] Wang Z, Gerstein M, Snyder M. RNA-Seq: a revolutionary tool for transcriptomics. Nat Rev Genet, 2009, 10: 57–63
[25] Trapnell C, Hendrickson D G, Sauvageau M, Goff L, Rinn J, Pachter L. Differential analysis of gene regulation at transcript resolution with RNA-seq. Nat Biotechnol, 2013, 31: 46–53
[26] Costa V, Angelini C, De Feis I, Ciccodicola A. Uncovering the complexity of transcriptomes with RNA-Seq. J Biomed Biotechnol, 2010, 2010: 853916
[27] Murashige T, Skoog F. A revised medium for rapid growth and bio assays with tobacco tissue cultures. Physiol Plant, 1962, 15: 473–497
[28] Aken B L, Ayling S, Barrell D, Clarke L, Curwen V, Fairley S, Fernandez Banet J, Billis K, García Girón C, Hourlier T, Howe K, Hähäri A, Kokocinski F, Martin F J, Murphy D N, Nag R, Ruffier M, Schuster M, Tang Y A, Vogel J H, White S, Zadissa A, Flicek P, Searle S M. The Ensembl gene annotation system. Database (Oxford), 2016, 2016: baw093
[29] Trapnell C, Pachter L, Salzberg S L. TopHat: discovering splice junctions with RNA-seq. Bioinformatics, 2009, 25: 1105–1111
[30] Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley D R, Pimentel H, Salzberg S, Rinn J, Pachter L. Differential gene and transcript expression analysis of RNA—seq experiments with TopHat and Cufflinks. Nat Protocols, 2012, 7: 562–578
[31] Anders S, Pyl P T, Huber W. HTSeq: a Python framework to work with high-throughput sequencing data. Bioinformatics, 2015, 31: 166–169
[32] Love M I, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol, 2014, 15: 550
[33] Wang L, Feng Z, Wang X, Wang X, Zhang X. DEGseq: an R package for identifying differentially expressed genes from RNA-seq data. Bioinformatics, 2010, 26: 136–138
[34] Ashburner M, Ball C A, Blake J A, Botstein D, Butler H, Cherry JM, Davis A P, Dolinski K, Dwight S S, Eppig J T, Harris M A, Hill D P, Issel-Tarver L, Kasarskis A, Lewis S, Matese J C, Richardson J E, Ringwald M, Rubin G M, Sherlock G. Gene ontology: tool for the unification of biology. Nat Genet, 2000, 25: 25–29
[35] Kanehisa M, Goto S. KEGG: Kyoto encyclopedia of genes and genomes. Nucl Acids Res, 2000, 28: 27–30
[36] Kanehisa M, Goto S, Sato Y, Kawashima M, Furumichi M, Tanabe M. Data, information, knowledge and principle: back to metabolism in KEGG. Nucl Acids Res, 2014, 42 (database issue): D199–D205
[37] Jenuwein T, Allis C D. Translating the histone code. Science, 2001, 293: 1074–1080
[38] Kouzarides T. Chromatin modifications and their function. Cell, 2007, 128: 693–705
[39] Baumann K. Genome stability: cyclin’on mRNA. Nat Rev Mol Cell Biol, 2016, 17: 676–677
[40] Maga G, Hübscher U. Proliferating cell nuclear antigen (PCNA): a dancer with many partners. J Cell Sci, 2003, 116: 3051–3060
[41] Georgescu R, Langston L, O’Donnell M. A proposal: Evolution of PCNA’s role as a marker of newly replicated DNA. DNA Repair, 2015, 29: 4–15
[42] Strzalka W, Ziemienowicz A. Proliferating cell nuclear antigen (PCNA): a key factor in DNA replication and cell cycle regulation. Ann Bot, 2011, 107: 1127–1140
[43] Labib K, Tercero J A, Diffley J F X. Uninterrupted MCM2-7 function required for DNA replication fork progression. Science, 2000, 288: 1643–1647
[44] Wei L, Zhao X. A new MCM modification cycle regulates DNA replication initiation. Nat Struct Mol Biol, 2016, 23: 209–216
[45] Helariutta Y, Fukaki H, Wysocka-Diller J, Nakajima K, Jung J, Sena G, Hauser M, Benfey P N. The SHORT-ROOT gene controls radial patterning of the Arabidopsis root through radial signaling. Cell, 2000, 101: 555–567
[46] Grimplet J, Agudelo-Romero P, Teixeira R T, Teixeira R T, Martinez-Zapater J M, Fortes A M. Structural and functional analysis of the GRAS gene family in grapevine indicates a role of GRAS proteins in the control of development and stress responses. Front Plant Sci, 2016, 7: 353
[47] Woodward A W, Bartel B. Auxin: regulation, action, and interaction. Ann Bot, 2005, 95: 707–735
[48] Zhao Y. Auxin biosynthesis and its role in plant development. Annu Rev Plant Biol, 2010, 61: 49–64
[49] Abel S, Theologis A. Early genes and auxin action. Plant Physiol, 1996, 111: 9–17
[50] Rivas-San Vicente M, Plasencia J. Salicylic acid beyond defence: its role in plant growth and development. J Exp Bot, 2011, 62: 3321–3338
[51] Janda M, Ruelland E. Magical mystery tour: salicylic acid signalling. Environ Exp Bot, 2015, 114: 117–128
[52] Johnson C, Boden E, Arias J. Salicylic acid and NPR1 induce the recruitment of trans-activating TGA factors to a defense gene promoter in Arabidopsis. Plant Cell, 2003, 15: 1846–1858
[53] Bernhardt R. Cytochromes P450 as versatile biocatalysts. J Biotechnol, 2006, 124: 128–145
Transcriptome Analysis of Promotive Effects of Active Carbon on Growth and Development of Maize Seedlings from in vitro Cultured Immature Embryos
WANG Jin-Ping1,2, SUN Guo-Zhong2,*, and WANG Hai-Bo2,*
1College of Agronomy, Agricultural University of Hebei, Baoding 071001, China;2Institute of Genetics and Physiology, Hebei Academy of Agriculture and Forestry Sciences / Plant Genetic Engineering Center of Hebei Province, Shijiazhuang 050051, China
Immature embryos from the maize inbred line Chang 7-2 were collected at 14 days after pollination, and cultured on MS or MSA medium (MS medium plus active carbon) for nine days at 24°C. Active carbon significantly accelerated the growth and development of maize seedlings from cultured immature embryos. Using RNA-seq technique, the genes involved in the growth promotive effects of active carbon were analyzed. The presence of active carbon in the medium affected the gene expression in seedlings significantly. Number of up- and down-regulated genes was 1612 and 530 in roots, as well as 69 and 78 in shoots, respectively; indicating that active carbon mainly affects gene expression in roots. GO enrichment analysis showed that differentially expressed genes (DEGs) in roots were mainly involved in DNA packaging, DNA packaging complex and hydrolase activity; the DEGs in shoots were mainly involved in lipid metabolic process, extracellular region and peroxidase activity. The KEGG enrichment analysis showed that the DEGs in roots were significantly associated with energy metabolism, carbohydrate metabolism, lipid metabolism, amino acid metabolism, cell cycle and plant hormone signal transduction. The DEGs in shoots were significantly associated with biosynthesis of ubiquinone and other terpenoid-quinone compounds. Several key genes involved in the cell cycle pathway (i.e., CYC, CDH1, MCM3, PCNA2, and BUB1), signal transduction of auxin (Aux/IAA) and cytochrome function (CYP450 oxidase) were significantly up-regulated by active carbon. Ten DEGs were confirmed by Real-time quantitative PCR assay, suggesting that our data and analysis of transcriptome sequencing are reliable.
Maize; Immature embryo; Active carbon; Transcriptome
(
): 2017-03-21; Accepted(接受日期): 2017-04-20; Published online(网络出版日期): 2017-05-08.
10.3724/SP.J.1006.2017.01489
本研究由河北省财政专项(2009055001, F15R25)资助。
This study was supported by the Financial Fund Program of Hebei Province (2009055001, F15R25).
*通讯作者(Corresponding authors): 孙果忠, E-mail: 13933023804@163.com; 王海波, E-mail: nkywanghb@163.com第一作者联系方式: E-mail: 15226506557@163.com
URL: http://kns.cnki.net/kcms/detail/11.1809.S.20170508.1007.012.html
