采用聚合酶链式反应-变性梯度凝胶电泳技术分析小熊猫胃肠道菌群的多样性
2017-09-16邓家波牛李丽余建秋熊律晨倪学勤
李 杨 邓家波 牛李丽 余建秋 曾 燕 刘 倩 周 毅 熊律晨 倪学勤** 曾 东**
(1.四川农业大学动物医学院动物微生态研究中心,成都611130;2.成都动物园,成都610081)
采用聚合酶链式反应-变性梯度凝胶电泳技术分析小熊猫胃肠道菌群的多样性
李 杨1邓家波2*牛李丽2余建秋2曾 燕1刘 倩1周 毅1熊律晨1倪学勤1**曾 东1**
(1.四川农业大学动物医学院动物微生态研究中心,成都611130;2.成都动物园,成都610081)
本试验旨在研究小熊猫胃肠道菌群多样性。采用聚合酶链式反应(PCR)-变性梯度凝胶电泳(DGGE)技术结合条带的克隆测序、聚类分析和主成分分析(PCA)检测小熊猫胃肠道菌群结构及多样性。结果表明:1)PCR-DGGE图谱显示小熊猫胃肠道中有大量菌群,且不同部位的菌群结构存在一定的差异,相邻肠段菌群结构具有一定的相似性。结肠和粪便样品的菌群多样性较高,其次为胃和直肠样品,而空肠和回肠样品菌群多样性较低。2)小熊猫胃肠道菌群PCR-DGGE图谱中测序的条带大多数归为厚壁菌门(Firmicutes)、拟杆菌门(Bacteroides)、变形菌门(Proteobacteria)和疣微菌门(Verrucomicrobia),共性条带主要是未培养拟杆菌门细菌(uncultured Bacteroidetes bacterium)、粪肠球菌(Enterococcusfaecalis)、未培养梭状芽孢杆菌(unculturedClostridiumsp.)、乳酸乳球菌(Lactococcuslactis)和食窦魏斯氏菌(Weissellacibaria),其中厚壁菌门为优势菌群;特异性条带主要是丛毛单胞菌(Comamonassp.)、梭菌属(Clostridiumsp.)和Akkermansia。由此可见,小熊猫胃肠道中栖息着大量菌群,且其多样性按照胃肠道由前至后的顺序呈现高-低-高的趋势。
小熊猫;胃肠道菌群;PCR-DGGE;多样性;克隆测序
小熊猫(Ailrususfulgens)是喜马拉雅-横断山脉特有的小型珍稀野生动物,仅分布于中国、尼泊尔、不丹、锡金、印度和缅甸北部地区,被列为国家二级保护动物。小熊猫是食肉目浣熊科,但具有与大熊猫极为相似的地理分布、生理结构和行为生态,是高度特化的素食性食肉目动物,在分类、生态学等领域具有较高的科学研究价值[1]。但由于人类活动和栖息地的破坏,我国小熊猫的数量在过去的半个世纪已降低40%[2]。1936年我国动物园开始圈养和保护小熊猫,且随着2003年小熊猫捕捉许可证的禁止发放及科学合理的保护,其繁殖和种群得到提高。近年来,越来越多的研究表明,动物的生理代谢和生长发育与其胃肠道的微生物密切相关[3]。同样,小熊猫的肠道微生物在竹子的消化过程中扮演着重要角色[4]。目前,小熊猫的粪便菌群结构及组成等信息已初步被报道和研究[4-6],但其整个胃肠道菌群结构和组成的研究还未见报道,其中栖息的微生物及微生物的作用等信息大多仍处于未知状态。目前,对野生动物胃肠道微生物的研究和利用已成为其繁殖及种群保护的重要措施[7]。因此,胃肠道菌群结构和组成的研究对小熊猫的饲养、疾病预防及种群保护具有重要的指导意义。
现代分子生物学技术为肠道菌群结构的研究提供了强有力的保障,其中,聚合酶链式反应(PCR)-变性梯度凝胶电泳(DGGE)技术作为一种快速可靠的比较菌群多样性的指纹图谱技术,自1993年被Muyzer等[8]首次报道后,已被广泛应用于动物胃肠道菌群多样性的研究。因此,本研究采用PCR-DGGE技术结合条带的克隆测序,分析小熊猫胃肠道菌群结构及组成,为小熊猫的饲养管理、疾病预防及种群保护提供现实指导。
1 材料与方法
1.1试验材料
1.1.1 样品采集
1.1.2 试验试剂与仪器
1.2胃肠道细菌总DNA的提取
1.3细菌总DNA的16SrDNAV3区PCR扩增
使用大肠杆菌通用引物[8]对细菌的16S rDNA V3区序列进行PCR扩增。上游引物:5′-CGCCCGCCGCGCGCGGCGGGCGGGGCGGGGG
以Folin-酚试剂法测定蛋白质含量实验为例,将全班学生分成6~8组,每组按大纲要求自主查阅文献资料,制作PPT,教师提前发布关于原理和注意事项的课件以及微量移液器、721分光光度计的使用录像,感兴趣的学生也可参与实验准备。正式实验时,每组先由1名学生代表汇报,教师随时点评汇报内容和解答学生预习过程中的疑问,随后分别进行实验。如此,笔者在实验结束批改报告时发现,和以往相比,学生直接抄书的现象基本没有,实验讨论和结论部分的撰写不再敷衍,尽可能体现自己的见解和创新。
CACGGGGGGCCTACGGGAGGCAGCAG-3′(带下划线部分为“GC”夹子);下游引物5′-ATTACCGCGGCTGCTGG-3′。反应体系(25.0 μL):2×Taq Master Mix 12.5 μL,上、下游引物(10 μmol/L)各1.0 μL,模板DNA 1.0 μL,双蒸水(ddH2O)9.5 μL。反应条件:94 ℃ 4 min;94 ℃ 30 s,58 ℃ 30 s,72 ℃ 2 min,30个循环;72 ℃ 10 min。1.0%琼脂糖凝胶电泳检测PCR产物。
1.4PCR-DGGE凝胶电泳及条带克隆测序

1.5数据分析
采用Excel 2010、SPSS 19.0和NTSYS 2.1等软件对PCR-DGGE图谱进行菌群多样性分析、主成分分析(PCA)和聚类分析。
2 结果与分析
2.1小熊猫胃肠道菌群的PCR-DGGE图谱及聚类分析
PCR-DGGE图谱中,颜色深的条带代表优势菌群,条带的数量和位置分布代表细菌菌群的丰富度和种类。小熊猫胃肠道样品经PCR-DGGE检测分离出不同数量的条带,且条带的强度和位置存在一定差异(图1)。PCR-DGGE图谱显示,来自胃、十二指肠、空肠、回肠、结肠、直肠和粪便样品的条带数分别为15、10、6、13、19、16和17条;且胃肠道部位对菌群结构及组成影响较大,来自结肠和粪便样品的菌群多样性较高,其次为胃和直肠样品,而空肠和回肠样品菌群多样性较低。聚类分析(图1)表明,小熊猫十二指肠和回肠样品聚为一簇,其相似性系数较高,为0.76;来自结肠和直肠的样品聚为一簇,相似性系数为0.68;而来自胃和粪便的样品聚为一簇,相似性系数仅为0.53;小熊猫不同肠段样品与粪便和胃的样品分离开来,相似性系数仅为0.45,这表明小熊猫胃肠道不同部位菌群结构存在一定的差异性。
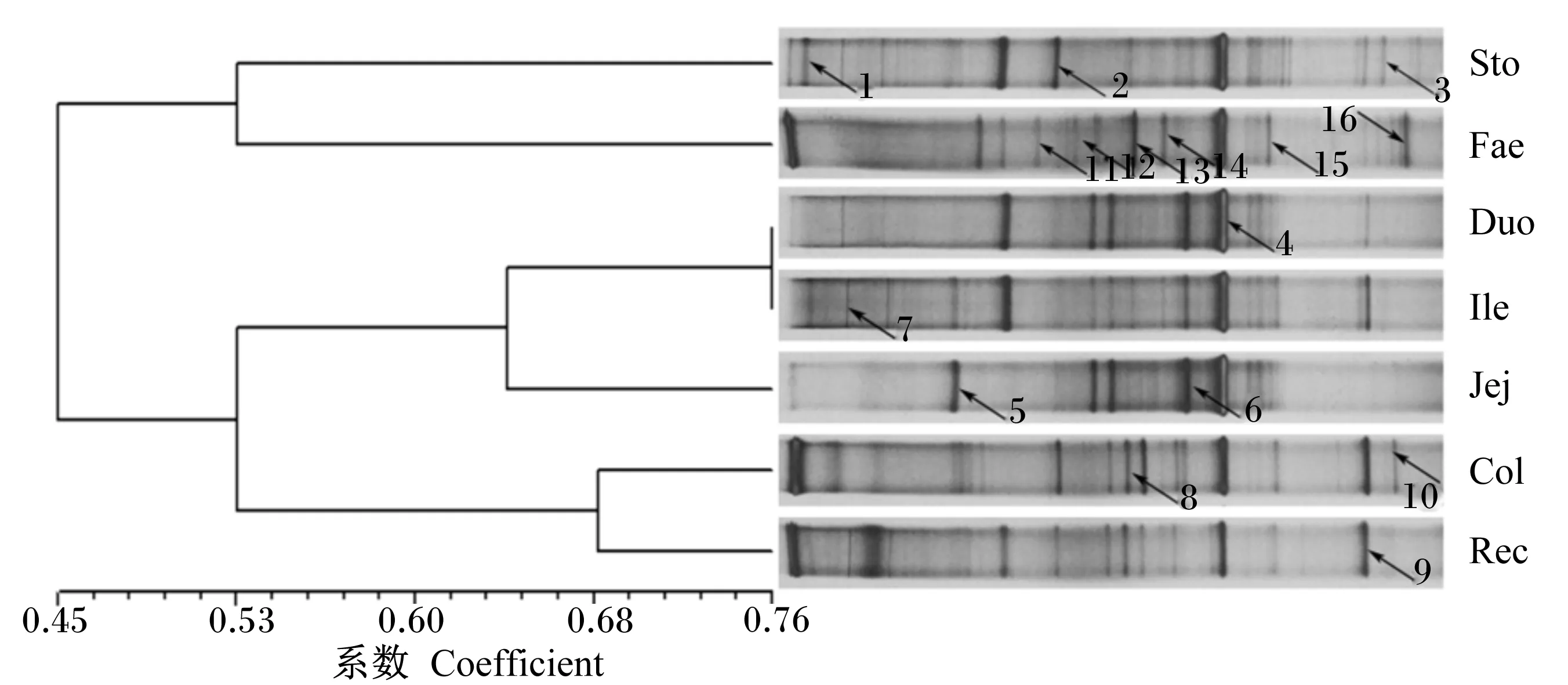
带箭头的1~16为条带编号。样品编号中Sto、Duo、Jej、Ile、Col、Rec和Fae分别表示胃、十二指肠、空肠、回肠、结肠、直肠和粪便样品。下图同。
1 to 16 with arrow are the bands No. Sto, Duo, Jej, Ile, Col, Rec and Fae stand for the samples from stomach, duodenum, jejunum, ileum, colon, rectum and faeces, respectively. The same as below.
图1小熊猫胃肠道菌群的PCR-DGGE图谱及聚类分析
Fig.1 PCR-DGGE profiles and cluster analysis of bacteria from gastrointestinal tract ofAilurusfulgens
2.2小熊猫胃肠道菌群PCR-DGGE图谱的多样性分析
由图2可知,小熊猫胃肠道不同部位菌群多样性存在差异,空肠样品菌群的香农多样性指数、均匀度和丰富度最低,分别为2.20、0.60和9;结肠样品菌群的多样性、均匀度和丰富度最高,分别为3.09、0.84和22;胃、直肠及粪便样品菌群的多样性、丰富度和均匀度较为相似,分别为2.89、0.78和18,2.94、0.80和19及3.00、0.81和20;按照胃肠道由前至后的顺序,肠道菌群多样性呈现高-低-高的趋势,这可能与胃肠道不同部位的消化功能及环境有关。因此,小熊猫的胃、十二指肠、空肠、回肠、结肠、直肠和粪便样品中,空肠样品菌群的多样性较低,结肠样品菌群的多样性较高,胃、直肠和粪便样品的菌群多样性较为相似,这与PCR-DGGE图谱分析结果一致。
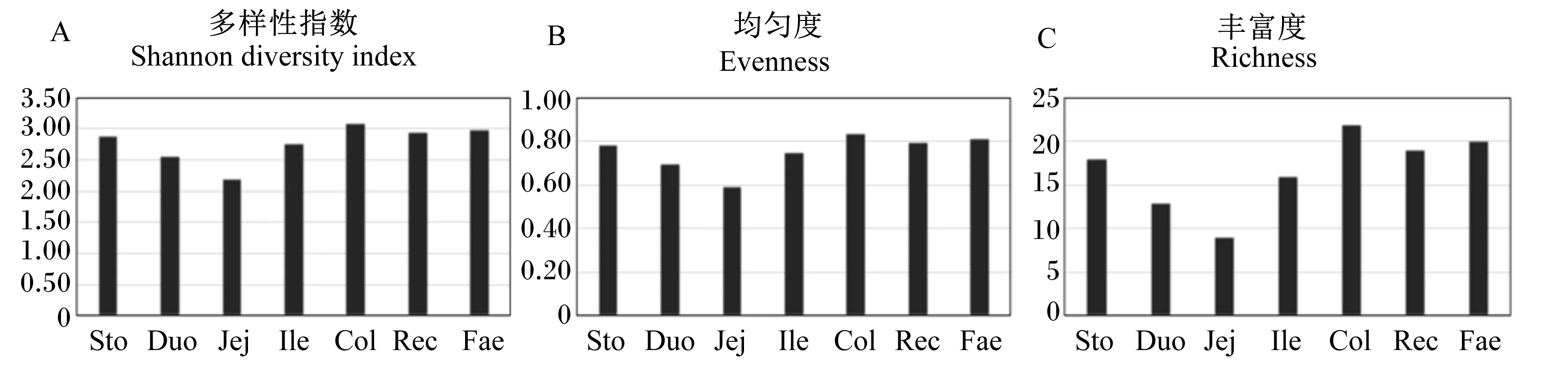
图2 小熊猫胃肠道菌群的香农多样性指数、均匀度及丰富度
2.3小熊猫胃肠道菌群PCR-DGGE图谱的PCA
PCR-DGGE图谱的PCA(图4)与聚类分析的结果一致。主成分因子1(PC1)的贡献率为33.56%,主成分因子2(PC2)的贡献率为24.70%。PC1明显将粪便样品与其他样品分开,而PC2明显将结肠、直肠和粪便样品与其他样品分开,表明小熊猫结肠、直肠和粪便样品菌群结构及组成与其他样品存在较大差异;而胃、十二指肠、空肠和回肠样品聚在一起,表明小熊猫胃肠道不同部位菌群结构及组成存在一定的差异性,但相邻部位样品的菌群结构及组成具有一定的相似性。
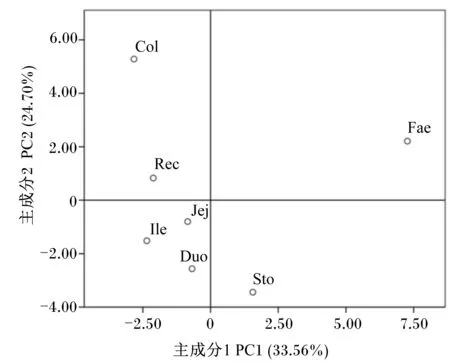
图3 PCR-DGGE图谱的PCA
2.4小熊猫胃肠道菌群PCR-DGGE图谱的共性条带和特异性条带分析
从PCR-DGGE图谱回收的16个条带(图1箭头所指)测序比对结果见表1。克隆测序结果显示(表2),小熊猫胃肠道的分离鉴定的共性条带主要来自厚壁菌门(Firmicutes)、拟杆菌门(Bacteroides)和变形菌门(Proteobacteria),包括大量未培养拟杆菌门细菌(uncultured Bacteroidetes bacterium)、粪肠球菌(Enterococcusfaecalis)、大肠杆菌(Escherichiacoli)、未培养细菌(uncultured bacterium)、约翰逊不动杆菌(Acinetobacterjohnsonii)、未培养梭状芽孢杆菌(unculturedClostridiumsp.)、乳酸乳球菌(Lactococcuslactis)和食窦魏斯氏菌(Weissellacibaria),其中有8株细菌属于厚壁菌门(占总分离鉴定菌的比例为50.00%),是小熊猫胃肠道中的优势菌群,肠球菌有3株(占总分离鉴定菌的比例为18.75%),是厚壁菌门中的优势菌群;特异性条带主要来自厚壁菌门、变形菌门和疣微菌门(Verrucomicrobia),分别是丛毛单胞菌(Comamonassp.)、布劳特氏菌(Blautiasp.)、未培养细菌、梭菌属(Clostridiumsp.)和Akkermansia。所有测序结果与GenBank数据库中微生物的相似性均不低于99%,说明试验中测序的序列与数据库中已鉴定的微生物的亲缘性较高。
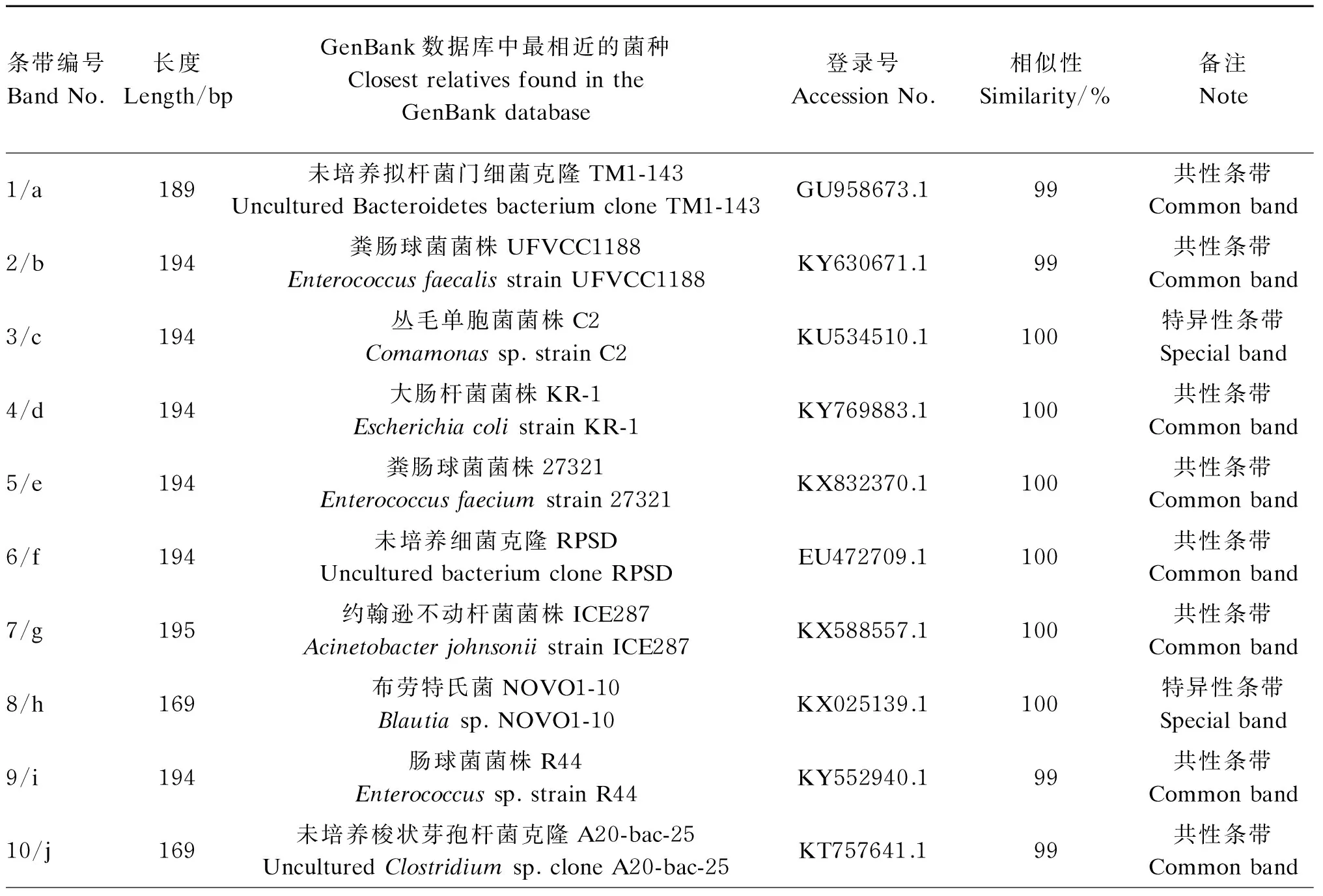
表1 PCR-DGGE图谱的共性条带和特异性条带的基因片段比对结果
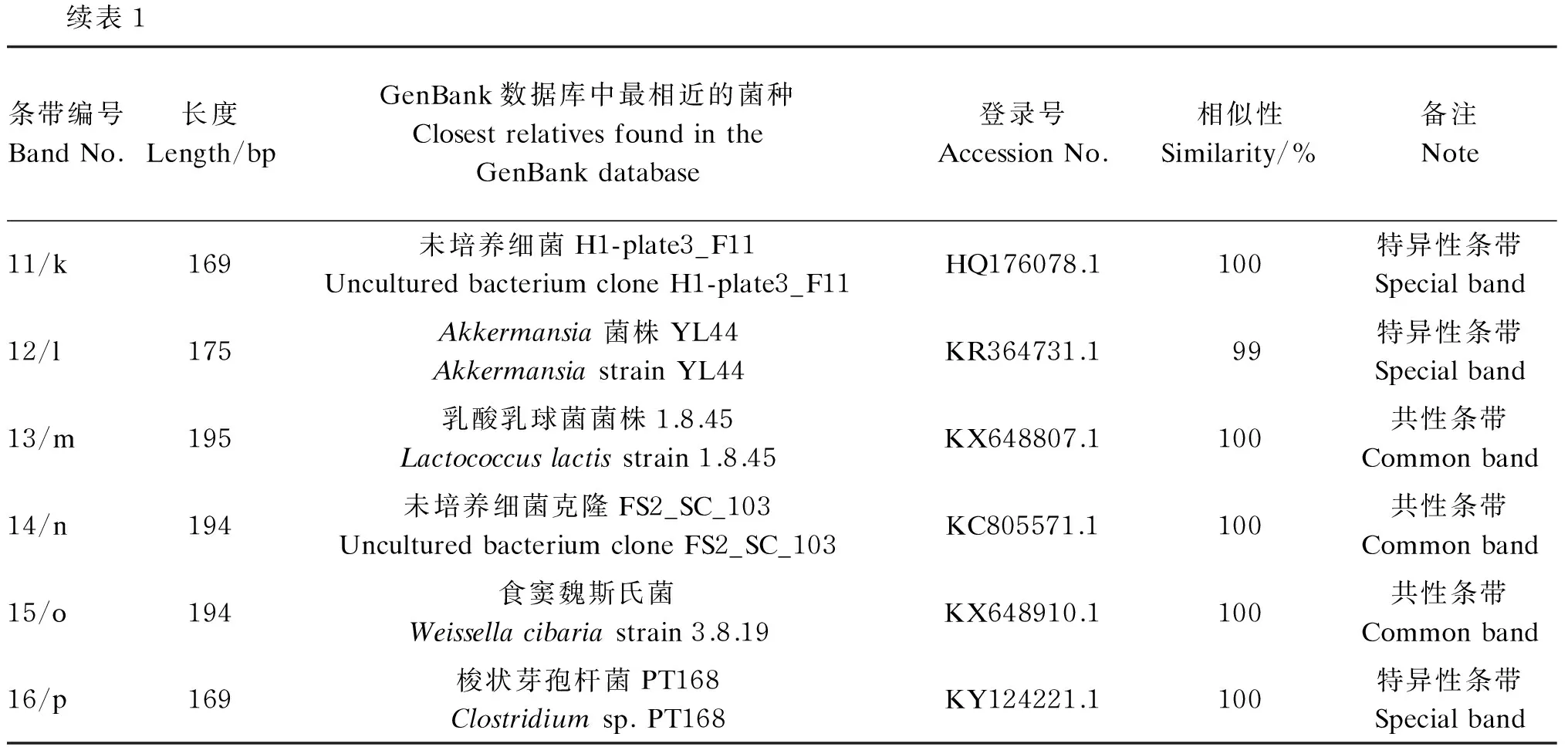
续表1条带编号BandNo.长度Length/bpGenBank数据库中最相近的菌种ClosestrelativesfoundintheGenBankdatabase登录号AccessionNo.相似性Similarity/%备注Note11/k169未培养细菌H1-plate3_F11UnculturedbacteriumcloneH1-plate3_F11HQ176078.1100特异性条带Specialband12/l175Akkermansia菌株YL44AkkermansiastrainYL44KR364731.199特异性条带Specialband13/m195乳酸乳球菌菌株1.8.45Lactococcuslactisstrain1.8.45KX648807.1100共性条带Commonband14/n194未培养细菌克隆FS2_SC_103UnculturedbacteriumcloneFS2_SC_103KC805571.1100共性条带Commonband15/o194食窦魏斯氏菌Weissellacibariastrain3.8.19KX648910.1100共性条带Commonband16/p169梭状芽孢杆菌PT168Clostridiumsp.PT168KY124221.1100特异性条带Specialband
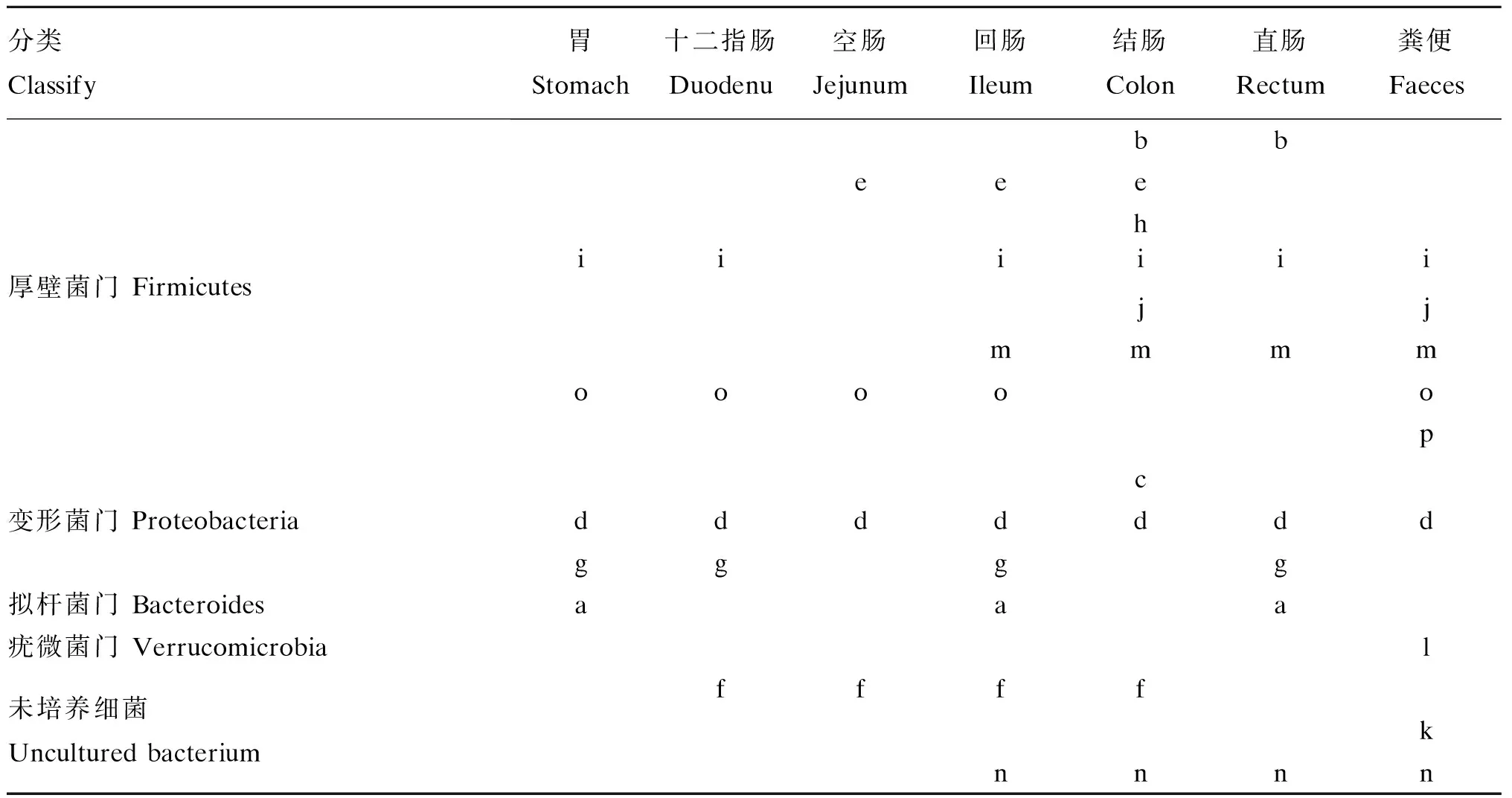
表2 克隆测序条带的分类及分布
表中字母为表1中条带编号 Letters in table were the band No. in Table 1。
3 讨 论
野生动物的种群繁殖及保护是目前全球科学家共同关注的核心问题[9],而其胃肠道微生物的研究和利用已成为其繁殖及种群保护的重要措施[7]。尽管小熊猫与大熊猫在分类上属于亲缘关系较远的2个物种,但全基因组的比较分析结果表明它们有与竹子生活方式相似的进化变化相关基因和酶类,使得它们适应低营养的竹子食物[1]。而胃肠道共生微生物与宿主的消化和生理功能密切相关[10]。因此,本研究分析了小熊猫胃、十二指肠、空肠、回肠、结肠、直肠和粪便样品PCR-DGGE图谱条带的分布、数量及强度等信息,在分子水平上快速、直观地反映了其胃肠道菌群的结构特点及多样性。PCR-DGGE图谱(图1)显示小熊猫胃肠道中存在着大量的菌群,而这些菌群与宿主的关系仍需进一步研究。尽管野生动物因其保护特性和生存特点而难以采集并对其胃肠道微生物进行深入研究,但近年来越来越多的野生动物胃肠道微生物信息被陆续报道[11-12],这些研究工作为全球野生动物的饲养管理和种群保护发挥了重要作用。
小熊猫胃肠道样品PCR-DGGE图谱的聚类分析结果(图1)显示,尽管十二指肠和回肠是非相邻的肠段,但其样品聚类在一起,且菌群结构及组成相似性较高,可能与小熊猫小肠结构特征及其食物的排空时间较短有关。小肠是消化吸收营养物质的主要部位,小熊猫的小肠比较发达,长度可达350 cm,且无盲肠[13],其消化道对食物的滞留时间在各种哺乳动物中最短[14]。PCR-DGGE图谱的PCA(图3)和菌群的多样性指数分析结果(图2)显示,小熊猫结肠样品菌群多样性较高,且按照胃肠道由前至后的顺序,样品菌群的多样性呈现高-低-高的趋势,这可能与胃肠道不同部位的消化功能及高纤维饮食有关[13,15]。研究报道,动物肠道部位对菌群结构和多样性有较大影响[12,16]。本研究中,小熊猫胃和粪便样品与其他肠段样品分离开来。小熊猫胃的容量大,且胃上皮分泌功能较强,胃腺黏液细胞发达且胃壁肌层组织厚,这些都有利于小熊猫对竹子等高纤维饮食的消化和吸收[17]。因本研究中的小熊猫是成都动物园临床死亡的动物,并未及时收集到其粪便样品,而用同圈舍其他小熊猫粪便样品替代分析菌群,且粪便样品处于动物消化道的末端并暴露于环境中,因此其菌群结构及组成与胃肠道的其他部位具有一定差异[18-19]。尽管如此,动物个体间及个体内部的菌群结构研究表明,同品种动物个体内部菌群结构差异大于个体间[20],因此,同圈舍其他小熊猫的粪便样品在一定程度能展现本研究中临床死亡小熊猫的粪便菌群结构信息。
PCR-DGGE图谱条带的克隆测序结果(表1)显示,小熊猫胃肠道菌群主要来自厚壁菌门、拟杆菌门、变形杆菌门和疣微菌门,且优势菌群主要是来自厚壁菌门的粪肠球菌和食窦魏斯氏菌。其中,食窦魏斯氏菌可参与肠道中纤维性食物的消化和吸收[21];粪肠球菌是哺乳动物肠道中主要的益生菌群,但其部分菌株仍具有致病性,这依赖于其菌株毒力岛的检测[22]。同样,Williams等[6]利用16S焦磷酸测序研究大熊猫和小熊猫的粪便菌群,结果表明菌群主要来自厚壁菌门和变形菌门,且参与降解发酵纤维物质产生丁酸的梭菌属是小熊猫的主要优势菌群,其数量占75%;而Erysipelotrichaceae则是大熊猫的主要优势菌群,其数量占95%,这表明小熊猫和大熊猫利用不同的菌群进行纤维降解。而本研究中也仅在粪便中检测到梭菌属菌群,可能与小熊猫具有的无盲肠且结肠和直肠较短的大肠结构特点有关[23]。同样,Kong等[4]利用焦磷酸测序比较了野生和圈养小熊猫粪便菌群多样性,发现野生小熊猫肠道菌群多样性显著高于圈养小熊猫,主要以厚壁菌门为主,且粪便中大部分细菌具有降解纤维的功能,其在小熊猫消化竹子过程中起到重要作用。尽管如此,小熊猫胃肠道与纤维降解相关的菌群信息仍处于未知状态。本研究中,布劳特氏菌是小熊猫结肠中特有细菌,而结肠菌群在发酵饮食益生元生成短链脂肪酸及抗炎抗肿瘤中发挥重要作用[24]。而Akkermansia是小熊猫胃中特有细菌,该菌在改善肥胖和糖尿病方面具有重要作用[25],因此该菌可能在小熊猫发酵纤维饮食方面具有重要作用,但该作用有待进一步进行深入的研究和证实。而条件致病菌大肠杆菌和约翰逊不动杆菌分别在小熊猫胃和回肠中检测出,这2种菌在小熊猫健康状态不佳的情况下可能会导致其胃肠道疾病的发生。对临床感染犬瘟热病毒(CDV)大熊猫粪便菌群的研究发现,其感染CDV后肠杆菌科菌群数量增加[11]。
本研究应用PCR-DGGE技术首次直观的展现了1只临床死亡小熊猫胃肠道的菌群结构及组成,但仍有大量未培养菌群,且其菌群功能及与宿主的关系仍需进一步深入的研究。尽管如此,本研究结果仍可为小熊猫的饲养管理及疾病预防提供基础资料和现实指导。
4 结 论
① 小熊猫胃肠道中存在大量菌群且大部分与纤维消化相关,本研究分离鉴定的16株细菌主要来自厚壁菌门、拟杆菌门、变形杆菌门和疣微菌门,其中有8株细菌属于厚壁菌门(占总分离鉴定菌的比例为50.00%),是小熊猫胃肠道中的优势菌群,肠球菌有3株(占总分离鉴定菌的比例为18.75%),是厚壁菌门中的优势菌群;与纤维消化相关的菌群有梭菌属和食窦魏斯氏菌。
② 胃肠道不同部位菌群多样性存在差异,按照胃肠道由前至后的顺序,其多样性呈现高-低-高的趋势。
[1] HU Y,WU Q,MA S,et al.Comparative genomics reveals convergent evolution between the bamboo-eating giant and red pandas[J].Proceedings of the National Academy of Sciences of the United States of America,2017,114(5):1081-1086.
[2] WEI F W,FENG Z J,WANG Z W,et al.Current distribution,status and conservation of wild red pandasAilurusfulgensin China[J].Biological Conservation,1999,89(3):285-291.
[3] LEULIER F,MACNEIL L T,Lee W J,et al.Integrative physiology:at the crossroads of nutrition,microbiota,animal physiology,and human health[J].Cell Metabolism,2017,25(3):522-534.
[4] KONG F L,ZHAO J C,HAN S S,et al.Characterization of the gut microbiota in the red panda (Ailurusfulgens)[J].PLoS One,2014,9(2):e87885.
[5] LI Y,GUO W,HAN S S,et al.The evolution of the gut microbiota in the giant and the red pandas[J].Scientific Reports,2015,5:10185.
[6] WILLIAMS C L,SPARKS D L,KOUBA A J,et al.Giant and red pandas utilize distinct microbial communities for their bamboo diet degradation[C]//Proceedings of 2014 American Chemical Society.San Francisco,CA:American Chemical Society,2014.
[7] STUMPF R M,GOMEZ A,AMATO K R,et al.Microbiomes,metagenomics,and primate conservation:new strategies,tools,and applications[J].Biological Conservation,2016,199:56-66.
[8] MUYZER G,DE WAAL E C,UITTERLINDEN A G.Uitterlinden A G.Profiling of complex microbial populations by denaturing gradient gel electrophoresis analysis of polymerase chain reaction-amplified genes coding for 16S rRNA[J].Applied and Environmental Microbiology,1993,59(3):695-700.
[9] ESTRADA A,GARBER P A,RYLANDS A B,et al.Impending extinction crisis of the world’s primates:why primates matter[J].Science Advances,2017,3(1):e1600946.
[10] KRAUTKRAMER K A,KREZNAR J H,ROMANO K A,et al.Diet-microbiota interactions mediate global epigenetic programming in multiple host tissues[J].Molecular Cell,2016,64(5):982-992.
[11] NA Z,MENG L,JING L,et al.Impacts of canine distemper virus infection on the giant panda population from the perspective of gut microbiota[J].Scientific Reports,2017,7:39954.
[12] MCKENNEY E A,RODRIGO A,YODER A D.Patterns of gut bacterial colonization in three primate species[J].PLoS One,2015,10(5):e0124618.
[13] 张淑云,王玉玺.小熊猫消化系统解剖[J].野生动物学报,1986(6):49-50.
[14] 吴登虎,谢幼新,胡洪光,等.小熊猫的消化试验[J].西华师范大学学报:自然科学版,1995(4):336-342.
[15] AFRIN T,TSUZUKI T,KANWAR R K,et al.The origin of the antibacterial property of bamboo[J].Journal of the Textile Institute,2012,103(8):844-849.
[16] 曾燕,曾东,倪学勤,等.应用PCR-DGGE技术比较绵羊瘤胃、网胃、瓣胃和皱胃菌群的多样性[J].动物营养学报,2015,27(1):298-304.
[17] 米志平,杨智.小熊猫胃的解剖和组织结构研究[J].四川动物,2001,20(4):214-216.
[18] TANCA A,MANGHINA V,FRAUMENE C,et al.Metaproteogenomics reveals taxonomic and functional changes between cecal and fecal microbiota in mouse[J].Frontiers in Microbiology,2017,8:391-403.
[19] PAUWELS J,TAMINIAU B,JANSSENS G P J,et al.Cecal drop reflects the chickens’ cecal microbiome,fecal drop does not[J].Journal of Microbiological Methods,2015,117:164-170.
[20] ZHU A,SUNAGAWA S,MENDE D R,et al.Inter-individual differences in the gene content of human gut bacterial species[J].Genome Biology,2015,16(1):82.
[21] LEE K W,HAN N S,KIM J H,et al.Purification and characterization of beta-glucosidase fromWeissellacibaria37[J].Journal of Microbiology & Biotechnology,2012,22(12):1705-1713.
[22] BAKSHI U,SARKAR M,PAUL S,et al.Assessment of virulence potential of uncharacterizedEnterococcusfaecalisstrains using pan genomic approach-identification of pathogen-specific and habitat-specific genes[J].Scientific Reports,2016,6:38648.
[23] 孙克显,吴天祺,谈克毅,等.小猫熊消化系统的解剖观察[J].西北民族大学学报:自然科学版,1980(2):3-12.
[25] PLOVIER H,EEVERARD A,DRUART C,et al.A purified membrane protein fromAkkermansiamuciniphilaor the pasteurized bacterium improves metabolism in obese and diabetic mice[J].Nature Medicine,2016,23(1):107-113.
*Contributed equally
**Corresponding authors: NI Xueqin, professor, E-mail: xueqinni@foxmail.com; ZENG Dong, professor, E-mail: zend@sicau.edu.cn
(责任编辑 菅景颖)
Bacteria Diversity in Gastrointestinal Tract of Ailurus fulgens Analyzed by Polymerase Chain Reaction-Denaturing Gradient Gel Electrophoresis Technology
LI Yang1DENG Jiabo2*NIU Lili2YU Jianqiu2ZENG Yan1LIU Qian1ZHOU Yi1XIONG Lyuchen1NI Xueqin1**ZENG Dong1**
(1. Institute of Animal Microecology College of Veterinary Medicine, Sichuan Agricultural University, Chengdu 611130, China; 2. Chengdu Zoo, Chengdu 610081, China)
This study aimed to evaluate the bacterial diversity in the gastrointestinal tract (GIT) ofAilurusfulgens. The polymerase chain reaction (PCR)-denaturing gradient gel electrophoresis (DGGE) technology combined with the cloning sequencing, cluster analysis and principal component analysis (PCA) of bands were used to detect the bacterial composition and diversity. The results showed as follows: 1) numerous bacteria were obtained from the GIT ofAilurusfulgensby using the PCR-DGGE profiles. There was a difference in each intestinal segment. However, there were some similarities between the adjacent intestinal segment. Samples from the colon and faeces had the higher bacterial diversity, the secondly for samples from stomach and rectum, and the samples from the jejunum and ileum had the lower bacterial diversity. 2) The sequencing results of PCR-DGGE profiles of bacteria from gastrointestinal tract ofAilurusfulgensshowed that most of the bacteria belonged to Firmicutes, Bacteroides, Proteobacteria and Verrucomicrobia. The common bands were mainly inclued uncultured Bacteroidetes bacterium,Enterococcusfaecalis, unculturedClostridiumsp.,LactococcuslactisandWeissellacibaria, and Firmicutes was the dominant bacterial. The specific bands wereComamonassp.,Clostridiumsp. andAkkermansia. In summary, a large amounts of bacteria exist in the GIT ofAilurusfulgens, and the diversity of bacteria shows a tendency of high-low-high according to the GIT from front to back.[ChineseJournalofAnimalNutrition,2017,29(9):3167-3174]
Ailurusfulgens; gastrointestinal tract bacteria; PCR-DGGE; diversity; cloning sequencing
10.3969/j.issn.1006-267x.2017.09.019
2017-03-29
国家自然科学基金(31672318);成都大熊猫繁育研究基金会项目
李 杨(1992—),女,辽宁喀左人,硕士研究生,从事动物微生态研究。E-mail: liyang@ stu.sicau.edu.cn
:倪学勤,教授,博士生导师,E-mail: xueqinni@foxmail.com;曾 东,教授,博士生导师,E-mail:zend@sicau.edu.cn
S826
:A
:1006-267X(2017)09-3167-08
*同等贡献作者
**
