大黄素和大黄素甲醚抗氧化活性的密度泛函理论研究
2017-09-16王宜尚
曹 曼,王宜尚,陈 哲,裴 玲
(滨州学院 化学化工学院 滨州市材料化学重点实验室,山东 滨州 256603)
大黄素和大黄素甲醚抗氧化活性的密度泛函理论研究
曹 曼,王宜尚,陈 哲,裴 玲
(滨州学院 化学化工学院 滨州市材料化学重点实验室,山东 滨州 256603)
本文采用密度泛函理论(DFT)方法,在B3LYP/6-311G(d,p)水平下对气相和溶剂中大黄素和大黄素甲醚及其可能解离途径中形成的自由基进行结构优化,在结构优化的基础上用B3LYP/6-311++G(2 d,2 p)基组计算了其相应的单点能,通过三种抗氧化反应机制、清除·OOH自由基的机理等方面分析了分子活性位与其性质的关系,分析了两种物质酚羟基在不同环境中抗氧化活性的能力大小。计算结果表明,大黄素和大黄素甲醚分子分别在C3和C8位上酚羟基活性最高,是最大可能的活性位点,大黄素的酚羟基活性略高于大黄素甲醚。在气相中氢原子转移是主要的机制,在极性溶剂中单电子转移是主要机制,在非极性溶剂中连续的质子损失电子转移是主要机制。
活性位点;抽氢反应;酚羟基;键解离焓
大黄素和大黄素甲醚都属蒽醌类化合物,实验研究发现大黄素和大黄素甲醚具有抗氧化作用,能抑制过氧化物的形成和活性氧产生,清除并捕捉自由基,达到抗氧化性的目的[1-2]。而对大黄素和大黄素甲醚的研究主要关注在实验及作用层面上[3-4],对其各个酚羟基位点反应的抗氧化活性的作用机理报道较少。量子化学计算可以很好地预测药物分子的抗氧化活性的强弱[5-6],为了从理论上阐述大黄素和大黄素甲醚清除自由基活性能力的原理,本课题拟通过密度泛函理论的方法,从大黄素和大黄素甲醚不同位置上酚羟基清除自由基的活性方面进行研究,进而对其抗氧化活性进行理论评价。
1 计算方法
本研究采用密度泛函理论(DFT)的方法在B3LYP/6-311G(d,p)基组水平下,对大黄素和大黄素甲醚单体及其相应自由基进行结构优化,然后在B3LYP/6-311++G(2d,2p)基组水平下进行单点能计算。基于大黄素和大黄素甲醚分子的结构参数、三种反应机制[7-8]、与自由基的反应机理,详细分析两分子各个酚羟基清除自由基活性的强弱。溶剂中的计算采用CPCM 模型[9],所有的计算通过Gaussian 03程序完成[10]。
2 结果与讨论
2.1 三种反应机制分析
2.1.1 氢原子转移机制
氢原子转移(HAT)是物质抗氧化反应的一种很重要的反应机制,尤其是在非极性溶剂中,其反应机理为R·+ArOH→RH+ArO·,抗氧化剂(ArOH)与自由基(R)反应,由一个氢原子转移到R通过均裂键的断裂完成。ArOH的反应活性可以由酚羟基的键解离焓(BDE)值来估计,BDE值越低,清除自由基的活性越高,反应越容易进行[11]。BDE的计算公式为BDE=Hr+Hh-Hp,其中Hr、Hh、Hp分别代表化合物自由基的焓、氢原子的焓、化合物的焓[7-8]。
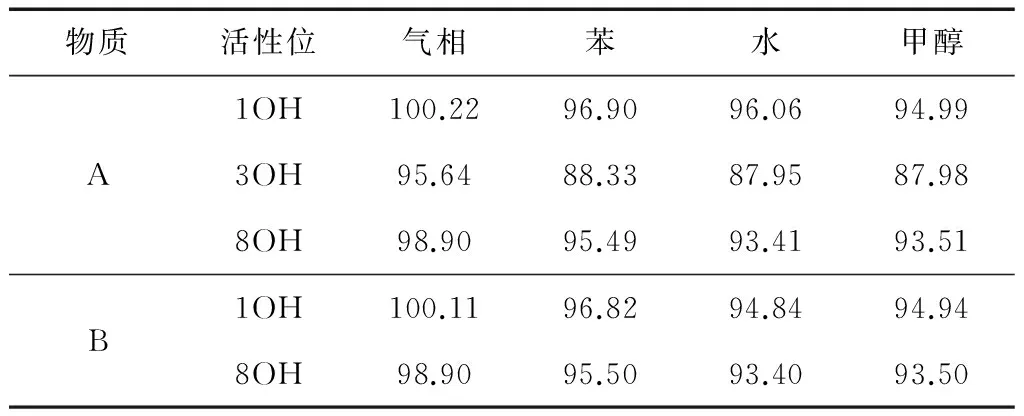
表1 大黄素和大黄素甲醚脱氢自由基在不同溶剂中的 键解离焓(BDE)(单位:kcal/mol)
BDE值越小,反应需要的热量越小,即反应越容易进行,酚羟基活性越大。从表1数据中可以看出无论是在哪种溶剂中大黄素C3位酚羟基的BDE值最小,大黄素和大黄素甲醚C8位酚羟基的BDE值都略小于C1位的,说明大黄素HAT反应的最大可能活性位点是C3位的酚羟基,两物质C8位酚羟基的活性略强于C1位的。比较溶剂对两物质不同位置酚羟基BDE的影响可以看出,对应酚羟基的BDE数值在苯非极性溶剂中略小于气相的,水和甲醇极性溶剂中又略小于苯中的,但整体来看溶剂对BDE数值影响不大,即HAT机制在各相中都可发生反应。比较两物质同位点酚羟基的BDE值看出大黄素的酚羟基BDE低于大黄素甲醚的,说明大黄素的抗氧化活性要强于大黄素甲醚。
2.1.2 单电子转移机制
单电子转移(SET-PT)机制反应式为R·+ArOH→R-+ArOH+·→RH+ArO·,此机制涉及到两个步骤:ArOH失去电子形成自由基正离子ArOH+,随后ArOH+失去H+成自由基ArO·,H+与R-形成RH。电离势(IP)主要反映分子的给电子能力,IP越小说明物质是优良的电子供体;质子解离焓(PDE)反映物质给出质子的能力,PDE值越小说明物质是优良的质子供体。在此两步反应中,IP和PDE可用作自由基清除活动评价中有效的能量因子,IP和PDE越小说明物质抗氧化性越强。IP与PDE计算公式为IP=Hcr-Hp+He, PDE=Hr+Hh+-Hcr,其中He、Hcr、Hh+分别代表电子的焓、阳离子自由基的焓、质子的焓[7-8]。
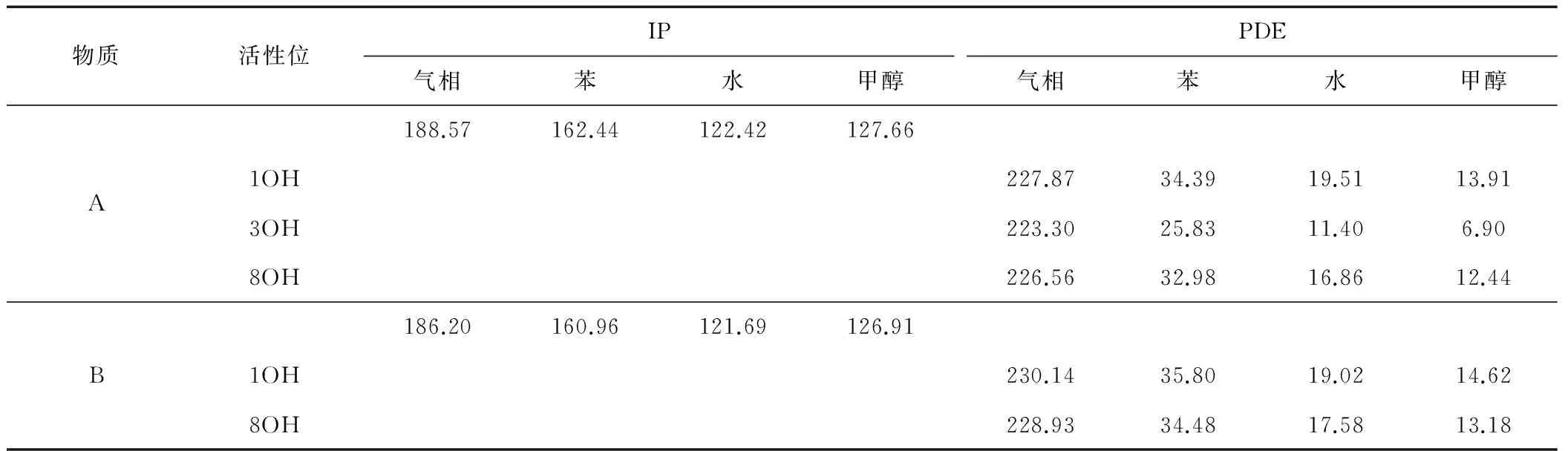
表2 大黄素和大黄素甲醚在不同溶剂中的电离势(IP)和质子解离焓(PDE)(单位:kcal/mol)
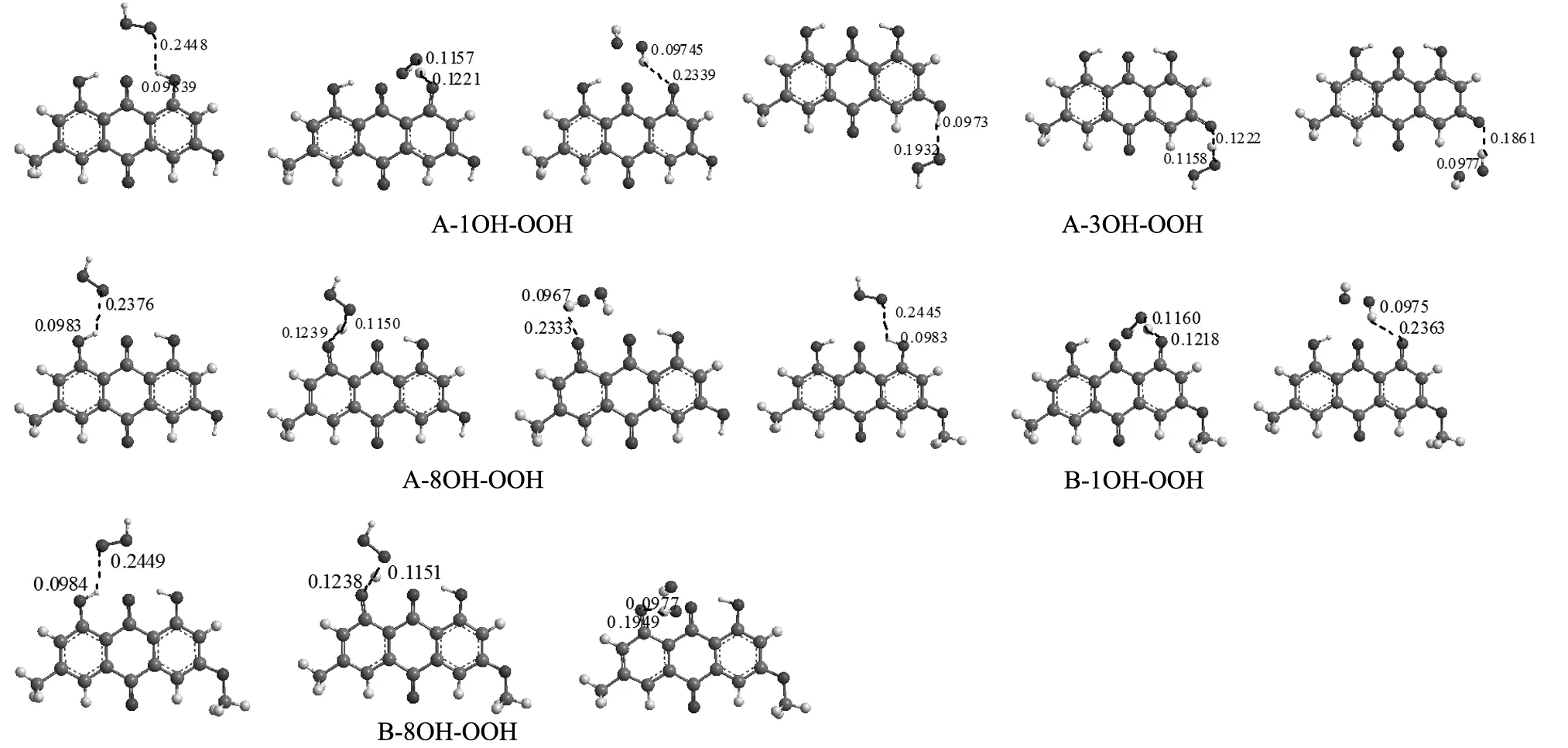
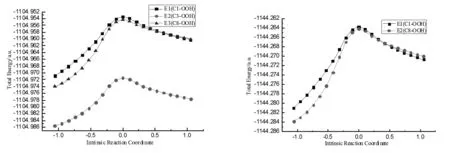
由表2中数据看出,两物质在同一溶剂中的IP数值差别不大,大黄素甲醚略小于大黄素的,说明在单电子转移过程中大黄素甲醚略优于大黄素。大黄素不同位置上脱氢自由基的PDE的大小顺序为C3 2.1.3 连续的质子损失电子转移机制 连续的质子损失电子转移(SPLET)机制为ArOH→ArO-+H+ArO-+R→ArO+R-R-+H+→RH ,首先ArOH失去去一个H+,形成ArO-,然后ArO-中的电子转移到自由基R上形成R-,最后R-与H+形成RH。由此看出,质子亲和能(PA)和电子转移焓(ETE)可以用来衡量抗氧化剂清除自由基活性的相对大小。PA和ETE的值越小说明清除自由基活性越大,PA、ETE的计算公式为PA=Han+Hh+,ETE=Hr+He-Han,其中Han、Hh+、Hp、Hr、He分别代表阴离子基团的焓、质子的焓、母体分子的焓、苯氧自由基的焓、电子的焓[7-8]。 表3 大黄素和大黄素甲醚在不同溶剂中的质子亲和能(PA)及电子转移焓(ETE)(单位:kcal/mol) 由表3数据可得在不同溶剂中大黄素PA的大小顺序为C3 2.2 大黄素和大黄素甲醚清除·OOH自由基的机理分析 ·OOH自由基为高活性自由基,单电子位于端位O原子,大黄素和大黄素甲醚能清除·OOH自由基,反应驻点物种结构图如图2所示。 图2 优化得到的反应物、过渡态、产物的几何结构(键长单位:nm) 为了确定所得为其真实的过渡态结构,在B3LYP/6-311G(d,p)基组水平上对每个过渡态进行了分子内反应坐标(IRC)计算。经过计算发现,IRC计算曲线两边显示的分子构型分别指向对应的反应物和产物,由此可见所得的过渡态结构为其真实过渡态,IRC曲线如图3所示。 图3 在B3LYP/6-311G(d,p)基组水平下计算得到的IRC曲线 如图2所示,当·OOH进攻大黄素C3位的酚羟基时,反应复合物O3…H3之间为正常的O-H键,H3…O之间的距离为0.1932 nm,接着O3…H3之间的距离逐渐増长为0.1222 nrn,H3…O之间的距离则缩短到0.1158nrn形成过渡态,经计算需越过37.386 kJ/mol的能垒,最后经过产物复合物形成醌式结构和H2O2,C3=O3之间为正常的C=O双键。·OOH与大黄素、大黄素甲醚分子C1、C8位酚羟基发生抽氢反应的过程中,产物与反应物能量差分别为27.940、36.786、26.839、36.209kJ/mol。比较发现,C3位上酚羟基与·OOH发生抽氢反应时达到过渡态时所需要的能垒小于C1、C8位酚羟基发生抽氢反应达到过渡态时所需要的能垒,且C3位酚羟基发生抽氢反应时的反应热小于C1和C8位的,因此与大黄素C1和C8位酚羟基相比,C3位上酚羟基更容易与·OOH发生抽氢反应,是最大可能的活性位点。 结合三种抗氧化反应机制分析,大黄素的C3位酚羟基是最大可能的活性位点,大黄素甲醚C8位酚羟基是最大可能的活性位点。在气相和非极性溶剂中,HAT机制起主要作用;在极性溶剂中SET-PT是主要的反应机制。大黄素的抗氧化性略强于大黄素甲醚。不同位置上酚羟基与·OOH发生抽氢反应时达到过渡态所需要的能垒C3< C1< C8,整个反应的反应热大小顺序也是C3< C1< C8,这都充分说明大黄素C3位、大黄素甲醚C1位酚羟基是最大可能的活性位点。 [1] 刘全德,唐仕荣,宋 慧,等.芦荟蒽醌类化合物的超声提取及其抗氧化性研究[J].食品与机械,2011,27(5):68-71. [2] 袁 晓,舒楚金,龚二兰,等.虎杖蒽醌化合物的分离及抗氧化活性的研究[J]. 食品研究与开发,2013,34(2): 22-24. [3] 潘晓丽,向 晖,谢运飞,等.大黄素-镁(Ⅱ) 配合物的合成、表征及抗氧化活性研究[J].中成药,2013,35(12):2757-2768. [4] 吕慧英,赵晨曦,吴 海,等.大黄提取物抗氧化活性与游离蒽醌相关性的研究[J].中草药,2010,41(3):412-415. [5] Ghiasi M, Majid M H.Quantum mechanical study of antioxidative ability and antioxidative mechanism of rutin (vitamin P) in solution[J]. Carbohydrate Research,2011,346:739-744. [6] María A M W, Sergio I C A, René R B Q. Computational study of the structure-free radical scavenging relationship of procyanidins[J]. Food Chemistry, 2014, 161:155-161. [7] Xue Y S,Zheng Y G,An L,et al.Density functional theory study of the structre -antioxidant activity of polyphenoli deoxybenzoins[J].Food Chemistry,2014,151:198-206. [8] Wang G R,Xue Y S.Theoretical study on the structural and antioxidant properties of some recently synthesised 2,4,5-trimethoxy chalcones[J].Food Chemistry,2015,171:89-97. [9] Cossi M,Barone V,Cammi R,et al.Ab initio study of solvated molecues:a new implementation of the polarizable continuum mode[J].Chemical Physics Letters,1996,255:327-335. [10] Fris ch M J,Trucks G W,Schlegel H B,et al.Gaussian03[M]. Gaussian Inc,Pittsburgh PA, 2003. [11] 张 军,陈德展.橄榄油中两种酚类化合物的抗氧化性理论研究[J].山东师范大学学报:自然科学版,2005,20(4):36-37. (本文文献格式:曹 曼,王宜尚,陈 哲,等.大黄素和大黄素甲醚抗氧化活性的密度泛函理论研究[J].山东化工,2017,46(3):14-16,18.) Density Functional Theory Study on the Antioxidant Activity of Emodin and Physcion CaoMan,WangYishang,ChenZhe,PeiLing (College of Chemistry &Chemical Engineering,Binzhou University,Binzhou Key Laboratory of Material Chemistry,Binzhou 256603,China) In this paper, the structure of emodin, physcion and their free radicals in the formation of dissociation pathways were optimized using density functional theory (DFT) method with the B3LYP/6-311G(d,p) basis set level in gas phase and solvents. Then with B3LYP/6-311++G(2d,2p) basis set level the corresponding single point energy were calculated. The activity of the phenolic hydroxyls on different sites of emodin and physcion was discussed on the basis of the structure parameters, the frontier molecular orbitals and three antioxidant reaction mechanisms. The results show that 3-OH and 8-OH are the most activist site, respectively. Emodin's antioxidant activity is higher than physcion. The results obtained also demonstrate that HAT would be the most favourable mechanism in the gas phase, whereas the SPLET mechanism is the thermodynamically preferred pathway in polar media, and SET-PT is the main mechanism in nonpolar media. active site;hydrogen abstraction reaction;phenolic hydroxyl;bond dissociation enthalpy 2016-11-18 曹 曼(1995—),女,山东临沂人,本科在读,研究方向:理论计算化学。 TQ461 A 1008-021X(2017)03-0014-03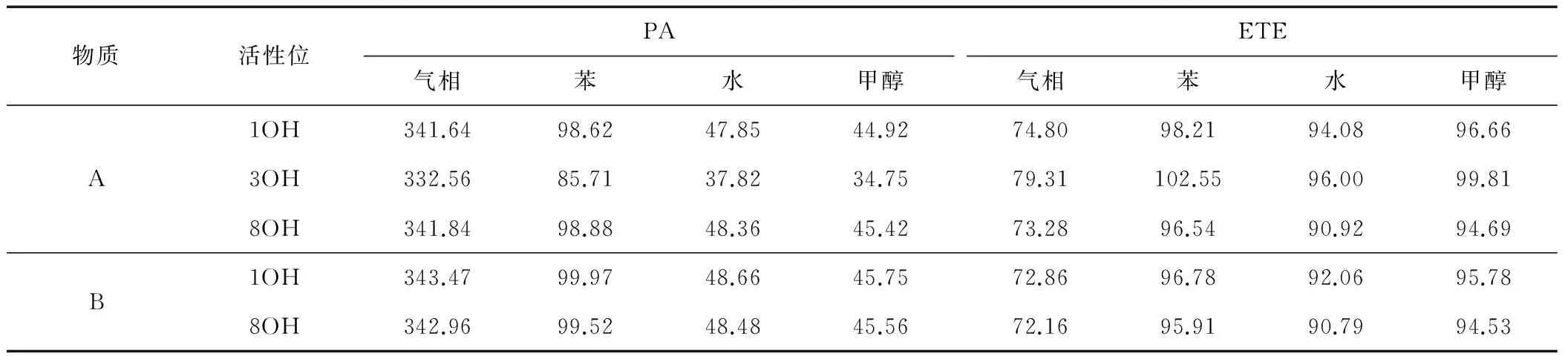
3 结论
