栽培稻×药用野生稻种间杂种基因组DNA甲基化的遗传与变异研究
2017-09-12蔡晓丹傅雪琳
何 平,疏 冕,蔡晓丹,傅雪琳
(1.华南农业大学 生命科学学院,广东 广州 510642;2.华南农业大学 农学院,广东 广州 510642)
栽培稻×药用野生稻种间杂种基因组DNA甲基化的遗传与变异研究
何 平1,疏 冕1,蔡晓丹1,傅雪琳2
(1.华南农业大学 生命科学学院,广东 广州 510642;2.华南农业大学 农学院,广东 广州 510642)
为了探索栽培稻与药用野生稻种间杂种体内异源基因组重组引起的DNA甲基化变异规律,及其生物学效应,利用甲基化敏感扩增多态性(MSAP)技术对辽粳944、药用野生稻及其杂种F1不同发育时期的叶片和小穗基因组DNA甲基化水平和模式的遗传变异特点进行了分析。结果表明,辽粳944、药用野生稻及其杂种F1的叶片和小穗DNA的甲基化水平表现出相同的变化趋势,即随着生长发育的进行,DNA甲基化水平呈下降趋势,各组织的全甲基化率均显著高于半甲基化率(P=0.000);杂种在分蘖期、减数分裂期及开花期叶片DNA平均总甲基化率、全甲基化率和半甲基化率分别为20.19%,16.06%及4.13%,其甲基化水平高于双亲;而杂种在减数分裂期、小孢子发育期及花粉成熟期小穗DNA平均总甲基化率、全甲基化率和半甲基化率分别为17.38%,13.67%及3.71%,均比辽粳944高,而比药用野生稻低;杂种小穗的半甲基化率显著高于辽粳944(P=0.015)。杂种叶片和小穗DNA的甲基化模式均有5种类型。经过对94个甲基化特异片段的序列分析,有38个片段的序列与已知或假定功能的基因具有同源性。为进一步研究栽培稻与野生稻种间杂种不育机理奠定了基础。
栽培稻;药用野生稻;种间杂种;DNA甲基化;甲基化敏感扩增多态性
药用野生稻(O.officinalis)属于CC染色体组,是原产我国的3种野生稻之一。药用野生稻在长期的自然适应与选择过程中积累了大量重要的有利基因,包括抗生物胁迫的防御基因、耐非生物胁迫的基因以及高光效基因等有利基因[1-2],这些基因多数都是目前水稻育种所重视和需要的。截至目前,栽培稻与野生稻种间杂交仍然是实现野生稻有利基因向栽培稻转移的主要途径,如在药用野生稻方面,研究者通过栽培稻与野生稻杂交和进一步的回交等,将药用野生稻的褐飞虱等的抗性基因导入到了栽培稻,获得了新的种质[3-4]。尽管如此,研究也表明,在AA组栽培稻与CC组药用野生稻种间杂交方面由于双方基因组的不同,存在着严重的交配障碍,其中杂种不育是影响基因交流和杂种利用的主要限制因素,对其机理的研究主要集中在减数分裂过程同源染色体配对及大孢子与小孢子发育的异常上,这种异常使得杂种表现为高度的雄性不育和雌性不育,进而影响结实[5]。研究也表明,即使在栽培稻背景下构建的药用野生稻单体异附加系的种质中,A和C 2个基因组间的互作显得非常复杂,影响了其遗传的稳定性和基因的表达[6],但是目前依然缺乏对其分子机理的研究。
众所周知,远缘杂交在作物种质创新和新品种培育上发挥着重要的作用,但远缘杂交后代会产生基因组重组或加倍形成异源多倍体,从而导致基因组变异和表型变异。由于远缘杂种体内高度异源的基因组的重组,产生细胞内染色体之间和质核之间的不协调,因此远缘杂交对植物基因组构成一种强烈冲击,在这种情况下,基因组内处于沉默状态的转座子(包括反转座子)可能被激活,从而对这一冲击发生应答并会引起基因组的重建即包括甲基化改变等的染色体结构性改变[7]。前人研究表明,远缘杂交后代F1及其染色体加倍形成异源多倍体的过程发生了广泛的甲基化变异。例如,在菰(ZizanialatifoliaGriseb)与栽培稻渐渗杂交系中发现,菰的DNA渗入引起了水稻全基因组和特异基因位点的甲基化变异[8];在小白菜品种矮抗青(B.rapa,AA)和芥蓝品种中华芥兰(B.oleracea,CC)杂交后代F1及其自交后代中,F1甲基化状态以双亲遗传为主,杂种F1甲基化状态发生变异的比例约为6%,同时13.1%的基因位点发生了甲基化修饰[9]。此外,在与植物生殖发育及育性有关的研究中,DNA甲基化也表现出变化,例如,Baroux等[10]对植物配子形成过程表观遗传与重编程的研究表明,植物配子形成过程中会发生一系列不同于亲本的包括甲基化和小分子RNA在内的调节变化;Ba等[11]在小麦雄性不育系与保持系的研究中,发现不同细胞质背景对核基因的甲基化影响表现出显著差异,与普通小麦遗传距离越远的供体种的细胞质产生的影响越大。Chen等[12]在玉米C型雄性可育和雄性不育杂种的研究中发现,育性恢复杂种的雄花甲基化水平显著高于雄性不育杂种,且在CMS-C育性恢复基因Rf5的区域发现一个特异的甲基化位点,表明DNA甲基化可能参与了CMS-C育性恢复基因的表达调控过程。由此看来,DNA甲基化作为最早发现的和最主要的表观遗传修饰方式之一,已被越来越多的研究发现其具有许多变异及重要的生物学功能。
水稻DNA甲基化的研究也在与之相关的多个方面都取得了重要进展,如抗性基因表达[13]、开花的基因控制[14]、转座子激活[15],DNA甲基化在水稻的生长、发育及基因表达调控等诸多过程中都发挥着极为重要的作用。更为重要的是,在水稻远缘杂交中产生的以DNA甲基化变异为主的表观遗传变异可能与杂种优势产生的分子机理密切相关,并在作物进化的过程当中起着十分关键的作用[16-18]。此外,Jin 等[19]在栽培稻与药用野生稻种间杂交研究中,对栽培稻与药用野生稻种间杂种F1(染色体组为AC)及其回交后代BC1F1(染色体组为AAC)抽穗期旗叶基因组DNA甲基化变异的研究表明,杂种F1的甲基化变异率接近7.6%,其中90.9%的甲基化位点变异在BC1F1得到保持,杂种基因和转座子都是甲基化变异发生的目标位点,且证明甲基化变异对杂种基因表达产生直接而快速的影响。尽管如此,至今未见有关栽培稻与野生稻种间杂种小穗发育过程中甲基化变异相关研究的报道,本研究利用甲基化敏感扩增多态性(MSAP)技术系统研究了辽粳944与药用野生稻杂种F1不同发育时期叶片及小穗基因组DNA甲基化的变异特点及相关基因表达,旨在为探讨栽培稻与药用野生稻种间杂种败育过程中的表观遗传修饰变异规律及其与杂种不育的关系提供参考。
1 材料和方法
1.1 供试材料
亲本材料分别为栽培稻品种辽粳944(O.sativa)(AA染色体组,2n=2x=24)、药用野生稻(O.officinalis)(CC染色体组,2n=2x=24,来源于中国广西),种间杂种为以辽粳944为母本、药用野生稻为父本进行杂交及杂种幼胚拯救所获得的杂种F1(AC染色体组,2n=2x=24)。各材料均盆栽种植于华南农业大学稻属核心种质收集圃。常规管理。
1.2 基因组DNA的提取
分别于分蘖盛期、小孢子母细胞减数分裂期和开花期取顶部第一片展开叶或剑叶叶片用于基因组总DNA的提取;此外,取每株主茎穗在减数分裂期、小孢子发育期及花粉成熟期小穗用于基因组总DNA的提取。每个材料各个发育时期提取DNA的叶片或小穗为3株相应组织的混合样品。DNA提取采用改良的CTAB法[20]。将提取出的DNA溶于TE并加入10 μL RNase(5 μg/μL)去除RNA,纯化后的DNA质量和浓度分别采用0.8%琼脂糖凝胶电泳和紫外分光光度计测定,于-20 ℃保存备用。
1.3 DNA甲基化敏感扩增多态性(MASP)分析
MSAP分析参考Xiong等[16]方法进行。具体如下:
1.3.1 接头序列、预扩增引物及选择性扩增引物(表1) MASP试验所用的EcoR Ⅰ、MspⅠ、HpaⅡ、TaqDNA聚合酶和T4连接酶等工具酶以及PCR反应体系均购自TaKaRa公司,凝胶回收试剂盒和质粒微量抽提试剂盒购自上海生工生物工程有限公司,总RNA抽提用TRIzol试剂盒购自Invitrogen公司,酵母提取物和蛋白胨购自Oxoid公司,其余试剂均为国产分析纯或化学纯试剂。
1.3.2 基因组DNA双酶切与连接 选取对甲基化敏感的同位酶MspⅠ、HapⅡ分别与限制性内切酶EcoR Ⅰ组成的双酶切组合EcoR Ⅰ/MspⅠ和EcoR Ⅰ/HpaⅡ进行酶切。30 μL酶切体系中含有10 μL基因组DNA(60 ng/μL),1 μLEcoR Ⅰ(15 U/μL),1 μLMspⅠ或HpaⅡ (10 U/μL),3 μL 10×T Buffer,3 μL 0.1% BSA,12 μL ddH2O。酶切体系置于37 ℃酶切5 h,70 ℃ 15 min,然后于-20 ℃保存备用或直接用于连接。30 μL连接体系含有酶切产物12.5 μL,50 μmol/LEcoR Ⅰ接头1和接头2各1 μL,50 μmol/LMspⅠ/HpaⅡ接头1和接头2各1 μL,T4连接酶1 μL (5 U/μL),10×T4Buffer 3 μL,ddH2O 9.5 μL。连接体系于4 ℃ 连接过夜。

表1 MSAP分析中的接头和扩增引物序列Tab.1 Sequences of adapters and primers used for MSAP analysis
1.3.3 连接产物预扩增 连接产物稀释10倍用于预扩增反应。30 μL预扩增体系含有稀释10倍的连接产物5.0 μL,预扩增引物1 和引物2 各1.5 μL,Taq酶0.3 μL (5 U/μL),10×PCR Buffer 3 μL,2.5 mmol/L dNTP 2.5 μL,ddH2O 16.2 μL。预扩增在EDC-810型基因扩增仪进行。扩增程序为:预变性94 ℃ 1 min;然后94 ℃ 30 s,56 ℃ 30 s,72 ℃ 60 s,30个循环;72 ℃ 10 min。反应结束后,取8 μL扩增产物于0.8%琼脂糖凝胶电泳检测,若产生均匀弥散的带型则符合条件,可用于下一步的选择性扩增反应,根据弥散的亮度确定稀释的倍数,通常稀释50倍。PCR产物 -20 ℃保存备用。
1.3.4 选择性扩增 选择性扩增体系30 μL,含有稀释50倍的预扩增产物5.0 μL,分别采用16种选择性引物组合,每个组合的成对扩增引物(表1中Primer 3~6,Primer 7~10)各1.5 μL,体系其余组分与预扩增体系相同。扩增程序为:94 ℃ 1 min;94 ℃ 30 s,65 ℃(每个循环递减1 ℃),72 ℃ 1 min,10个循环;94 ℃ 30 s,56 ℃ 30 s,72 ℃ 60 s,25个循环;72 ℃ 10 min。 PCR产物 -20 ℃保存备用。
1.3.5 电泳与银染检测 取选择性扩增产物8.0 μL,用6%聚丙烯酰胺凝胶电泳,银染显色,然后观察、记录并拍照。
1.3.6 MSAP 差异条带统计与甲基化位点分析 将在凝胶上显示清晰的电泳条带进行统计,对于各材料在同一位点有带则记为“+”,无带则记为“-”,记录所有引物组合的带型以及不同带型出现的数量,用于计算分析。具体如下:
甲基化敏感扩增多态性(MSAP)是在片段长度扩增多态性(AFLP) 技术的基础上经过改进的方法,常用到的2组双酶切组合为EcoR Ⅰ/MspⅠ、EcoR Ⅰ/HpaⅡ。MspⅠ和HpaⅡ都能识别CCGG序列,但是对胞嘧啶甲基化的敏感程度不同。MspⅠ对双链内甲基化(CmCGG)序列具有活性而对外甲基化(mCCGG) 序列敏感,HpaⅡ对半甲基化序列(即单链甲基化序列)具有活性而对全甲基化序列(即双链甲基化序列)敏感。对于基因组中任意一个CCGG位点,MSAP分析中EcoR Ⅰ/MspⅠ(E+M)和EcoR Ⅰ /HpaⅡ(E+H)2种酶切组合中可能出现的谱带一共有4种类型:Ⅰ、E+M和E+H 2种酶切都有带,表明该CCGG位点为非甲基化位点(图1,类型A);Ⅱ、E+M酶切有带而E+H酶切无带,表明该CCGG位点胞嘧啶发生了双链内甲基化,即全甲基化(图1,类型B);Ⅲ、E+M酶切中无带而E+H酶切中有带,表明该CCGG位点胞嘧啶发生了半甲基化(图1,类型C);Ⅳ、E+M和E+H 2种酶切中都无带,可能是该CCGG位点胞嘧啶发生了双链外甲基化或内侧和外侧胞嘧啶同时甲基化,但2种酶所不能识别的前述甲基化类型出现的频率很低,因此当2种酶切均无带时,认为不存在CCGG位点[21]。

图1 MSAP分析中谱带的类型
1.3.7 甲基化水平的计算与统计分析 全甲基化位点数(条带数)占总扩增条带数的百分率为全甲基化百分率,用以表示全甲基化水平;半甲基化位点数(条带数)占总扩增条带数的百分率为半甲基化百分率,用以表示半甲基化水平;全甲基化位点数与半甲基化位点数之和为总的甲基化位点数,用以表示基因组总的甲基化水平。利用SPSS 18.0软件进行甲基化水平的方差分析、多重比较和相关分析等。
1.4 MSAP特异条带的回收、克隆及测序
用灭菌手术刀从MSAP胶上切取特异性的条带,放入1.5 mL离心管中,加入100 μL ddH2O,用灭菌的枪头捣烂后沸水煮10 min,12 000 r/min离心2 min,取6 μL上清液作为PCR的模板,扩增的引物及程序与原选择性扩增中的保持一致。所得的PCR产物进行2%琼脂糖凝胶电泳检测,如果是一条明亮的条带并且大小和变性聚丙烯酰胺凝胶电泳的结果一致,则使用凝胶回收试剂盒回收并纯化目的条带,用于后续试验。
1.5 甲基化特异片段的半定量RT-PCR表达分析
分别以提取的不同材料的总RNA为模板进行反转录合成cDNA第一链,再进行PCR扩增,引物见表2。反应体系与扩增程序同1.3.3。反应产物经1%琼脂糖凝胶电泳检测。
2 结果与分析
2.1 杂种F1叶片和小穗基因组DNA甲基化水平的变化
用16对引物组合对亲本及其杂种F1不同发育时期的叶片和小穗基因组DNA MSAP分析,得到亲本及杂种F1不同时期的甲基化水平如图2和表3所示。对叶片组织而言,辽粳944、药用野生稻及杂种F1分别检测到2 672,3 479和3 702个双酶切条带,观察到全甲基化和半甲基化2种甲基化类型。而总甲基化率、全甲基化率和半甲基化率在各材料表现出相同的变化趋势,即随着生长发育的进行,从分蘖期、减数分裂期到开花期叶片基因组DNA甲基化水平呈下降趋势(图2-A、C、E),且各材料全甲基化率均显著高于半甲基化率(P=0.000),说明双亲及杂种F1叶片基因组DNA的甲基化以全甲基化为主要形式;3个材料的总甲基化率、全甲基化率和半甲基化率的大小表现为杂种F1>药用野生稻>辽粳944,但材料间差异不显著,尽管如此,杂种F1叶片基因组DNA平均总甲基化率、全甲基化率和半甲基化率分别比栽培稻亲本辽粳944高11.02%,8.56%和21.71%,比药用野生稻分别高2.16%,1.75%和3.77%,说明杂种F1较辽粳944亲本发生了更大的基因组甲基化变异,尤其表现在半甲基化水平。相关分析表明(表4),杂种F1总甲基化率、全甲基化率和半甲基化率与2个亲本的相应结果呈正相关关系,其中杂种F1全甲基化率与亲本辽粳944和药用野生稻的相关系数分别为0.998和1.000,达极显著相关水平,预示着甲基化水平具有明显的遗传决定趋势。

表2 用于甲基化特异片段RT-PCR的引物Tab.2 Primers used in RT-PCR of specific MSAP segments
注:TL.分蘖期叶片;ML.减数分裂期叶片;FL.开花期叶片;MT.减数分裂期小穗;XT.小孢子时期小穗;FT.花粉成熟期小穗。表5-6、图6同。
Note:TL.Leaf at tillering stage;ML.Leaf at meiosis stage;FL.Leaf at flowering stage;MT.Spikelet at meiosis stage;XT.Spikelet at microspore stage;FT.Spikelet at pollen matured stage. The same as Tab.5-6,Fig.6.
对小穗组织而言,辽粳944、药用野生稻及杂种F1小穗DNA的MSAP分析中分别检测到3 621,4 218,4 501个双酶切条带,观察到全甲基化和半甲基化2种甲基化类型。3个材料总甲基化率、全甲基化率和半甲基化率表现出相同的变化趋势,即从减数分裂期、小孢子发育期到花粉成熟期的小穗基因组DNA甲基化水平随着发育时期的推移逐渐降低(图2-B、D、F),且杂种F1的甲基化水平总是介于双亲之间,表现为药用野生稻>杂种F1>辽粳944的变化趋势,其中杂种F1的平均总甲基化率、全甲基化率和半甲基化率分别为17.38%,13.67%及3.71%,相应比栽培稻亲本辽粳944分别高4.43%,1.49%和16.88%,比药用野生稻低2.23%,0.75%和7.32%(表3)。方差分析表明,总甲基化率和全甲基化率在材料之间差异不显著,只有半甲基化率在材料间差异显著(P=0.015),即杂种F1和药用野生稻的小穗半甲基化率无显著差异,但均显著高于辽粳944。以上结果说明杂种F1小穗半甲基化水平较栽培稻亲本辽粳944发生了更大的变异。相关分析表明(表4),杂种F1总甲基化率、全甲基化率与2个亲本呈正相关关系,其中叶片的全甲基化率与2个亲本均呈显著相关,相关系数分别为0.998和1.000;而杂种小穗总甲基化率、全甲基化率和半甲基化率仅与亲本辽粳944相关显著,相关系数均为1.000。
总体来看,杂种与亲本的叶片、小穗DNA甲基化水平都随着生长发育的进行呈降低趋势,叶片的平均甲基化水平高于小穗。与亲本比较而言,杂种F1叶片和小穗甲基化水平都发生了变异,但只有小穗半甲基化水平较母本辽粳944显著增高。所有供试材料小穗和叶片的平均总甲基化率、全甲基化率和半甲基化率之间呈显著正相关(P≤0.001),相关系数分别为0.930,0.970和0.857。
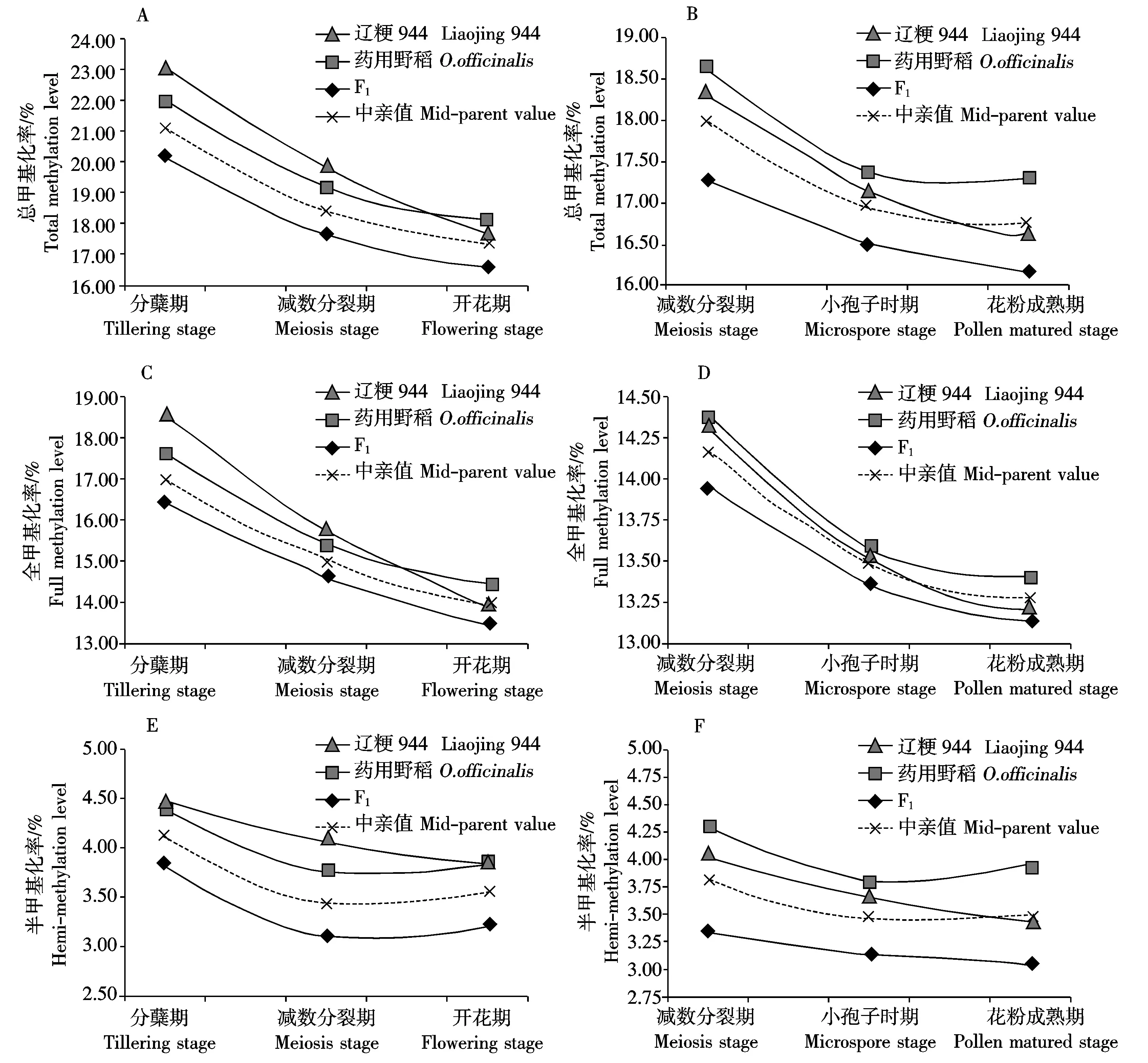
其中A、C、E图分别表示叶片DNA总甲基化率、全甲基化率和半甲基化率,B、D、F图分别表示小穗DNA总甲基化率、全甲基化率和半甲基化率。
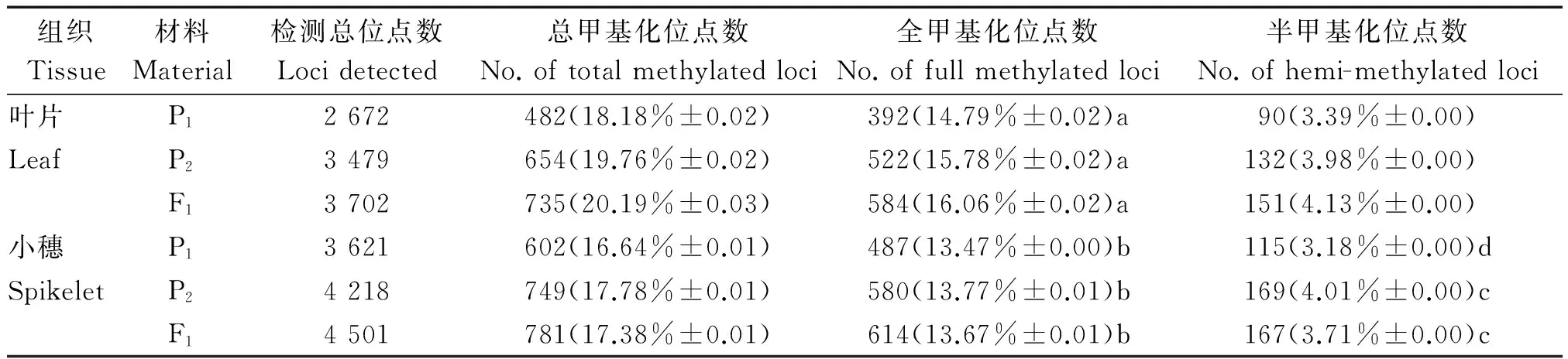
组织Tissue材料Material检测总位点数Locidetected总甲基化位点数No.oftotalmethylatedloci全甲基化位点数No.offullmethylatedloci半甲基化位点数No.ofhemi-methylatedloci叶片P12672482(18.18%±0.02)392(14.79%±0.02)a90(3.39%±0.00)LeafP23479654(19.76%±0.02)522(15.78%±0.02)a132(3.98%±0.00)F13702735(20.19%±0.03)584(16.06%±0.02)a151(4.13%±0.00)小穗P13621602(16.64%±0.01)487(13.47%±0.00)b115(3.18%±0.00)dSpikeletP24218749(17.78%±0.01)580(13.77%±0.01)b169(4.01%±0.00)cF14501781(17.38%±0.01)614(13.67%±0.01)b167(3.71%±0.00)c
注:P1.辽粳944;P2.药用野生稻。表4,5、图6同。括号外数字为甲基化位点数,括号内数值为甲基化率±标准误;a 表示同一材料叶片组织全甲基化水平显著高于半甲基化水平(P=0.000);b 表示同一材料小穗组织全甲基化水平显著高于半甲基化水平(P=0.000);c表示杂种F1与药用野生稻的小穗DNA半甲基化率差异不显著,但均显著高于辽粳944小穗(P=0.015)。
Note:P1.Liaojing 944,P2.O.officinalis. The same as Tab.4,5,Fig.6. The figures outside the brackets were number of methylated loci,values in brackets were methylation level±standard error;a means that the full methylation level in leaf tissue of the same material was significantly higher than the hemi-methylation level(P=0.000);b means that the full methylation level in spikelet tissue of the same material was significantly higher than the hemi-methylation level(P=0.000);c means that there was no significant difference of hemi-methylation level between the spikelets of hybrid F1andO.officinalis,but they were significantly higher than those of the Liaojing 944 spikelet (P=0.015).
2.2 杂种F1叶片和小穗基因组DNA甲基化模式的遗传与变异
通过与亲本甲基化位点的带型比较,杂种F1有A型、B型、C型、D型和和H型5种不同类型的甲基化位点模式(图3,4、表5 )。其中A型为单线态甲基化模式,即父本、母本和杂种F1在相同位点出现相同的甲基化条带。与单线态甲基化模式对应的为多线态甲基化模式,包括B型、C型、D型和H型4种类型,其中在B类型中,杂种F1在亲本甲基化位点的MSAP带型与亲本之一相同而与另一亲本不同,称为单亲甲基化遗传模式;在C类型中,亲本之一的甲基化条带在杂种F1中丢失,表明杂种F1在此位点发生了甲基化的变异,称为单亲甲基化变异模式;在D类型中,杂种F1的带型与2个亲本均不相同,表明杂种F1在此位点发生了甲基化变异,称为双亲甲基化变异模式;在H类型中,双亲无CCGG位点(2种酶切组合均无带)或其CCGG位点为非甲基化(2种酶切组合均有带),但杂种F1表现出了半甲基化或全甲基化模式,称为超甲基化变异。

表4 杂种F1与亲本甲基化率的相关系数Tab.4 Correlation coefficient of methylation levels between hybrid F1 and parents
注:TML.总甲基化率;FML.全甲基化 ;HML.半甲基化率。R指的是3个供试材料叶片和小穗甲基化率的相关系数。*和**分别表示在0.05和0.01 水平上显著相关。
Note:TML.Total methylation level;FML.Full methylation levl;HML.Hemi-methyation level.Rmeans the correlation coefficient of methylation levels between leaf and spikelet of all experimental materials.*and**present the significant correlation at 0.05 and 0.01 levels, respectively.
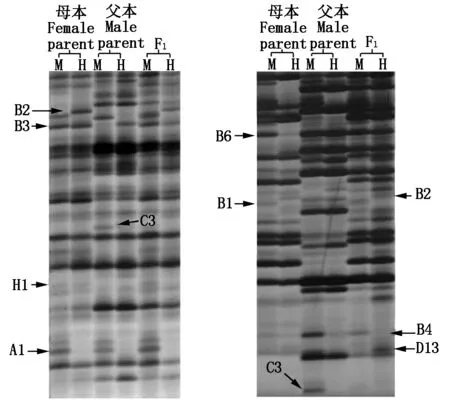
其中左图中引物组合为表1中primer3+primer8,右图中引物组合为primer3+primer7;M表示EcoRⅠ+MspⅠ酶切组合,H表示EcoR Ⅰ+HpaⅡ组合。
The primer combination in left and right figures are primer3+primer8 and primer3+primer7,respectively; M and H represent enzyme combinations ofEcoR Ⅰ+MspI andEcoR Ⅰ+HpaⅡ,respectively.
图3 亲本及杂种F1开花期旗叶在不同引物组合的甲基化电泳条带示例
Fig.3 A representative gel image of MSAP band of flag leaf at flowering stage
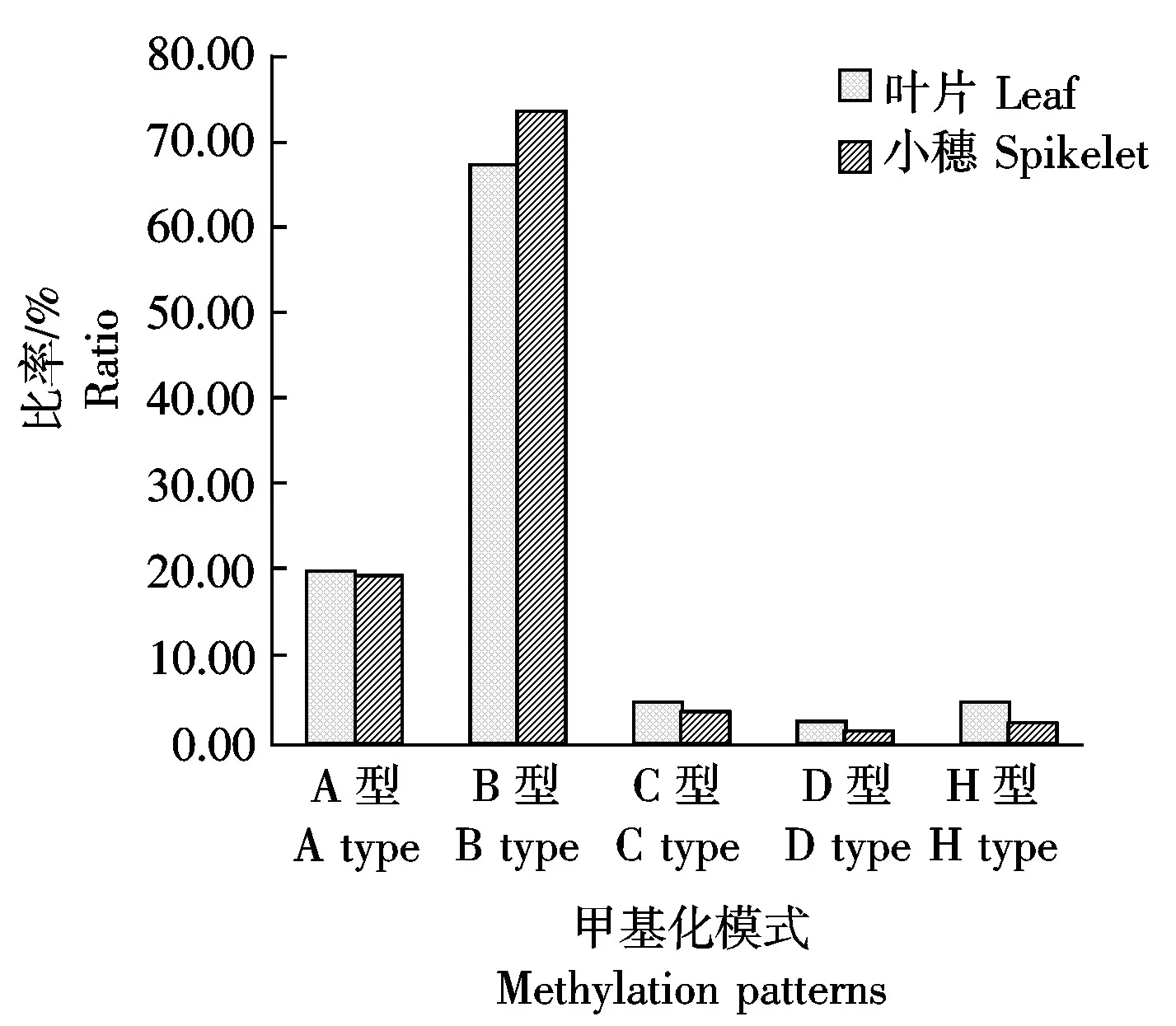
图4 杂种F1甲基化模式的类型与比率
结果表明,杂种F1叶片和小穗基因组DNA的甲基化模式均有以上5种类型,各时期5种甲基化类型的位点总数分别为911,1 053个,且各类型所占比率在2个组织中表现趋势相似,即均以B型为主要模式,占叶片和小穗甲基化位点总数的66.96% 和73.60%;其次是A型,即单线态甲基化模式,占叶片和小穗甲基化位点总数的20.20%和19.09%;C型、D型和H型只占叶片和小穗甲基化位点总数的1%~5%(表5、图4)。在A型和B型中,杂种F1叶片和小穗组织均以全甲基化模式为主要类型 (即表5中的A1、B1和B4亚类),与表3所示杂种的全甲基化水平显著高于半甲基化水平的结果一致。此外,杂种F1叶片在分蘖期、减数分裂期和开花期的基因组DNA甲基化模式的位点数分别为313,298和300个,分别占叶片DNA甲基化位点总数的34.36%,32.71%和32.93%;小穗在减数分裂期、小孢子发育期和花粉成熟期的甲基化位点数分别为343,349和361个,分别占小穗DNA甲基化位点总数的32.57%,33.14%和34.28%。以上结果表明,杂种F1DNA甲基化模式受组织器官和发育时期的影响非常小。
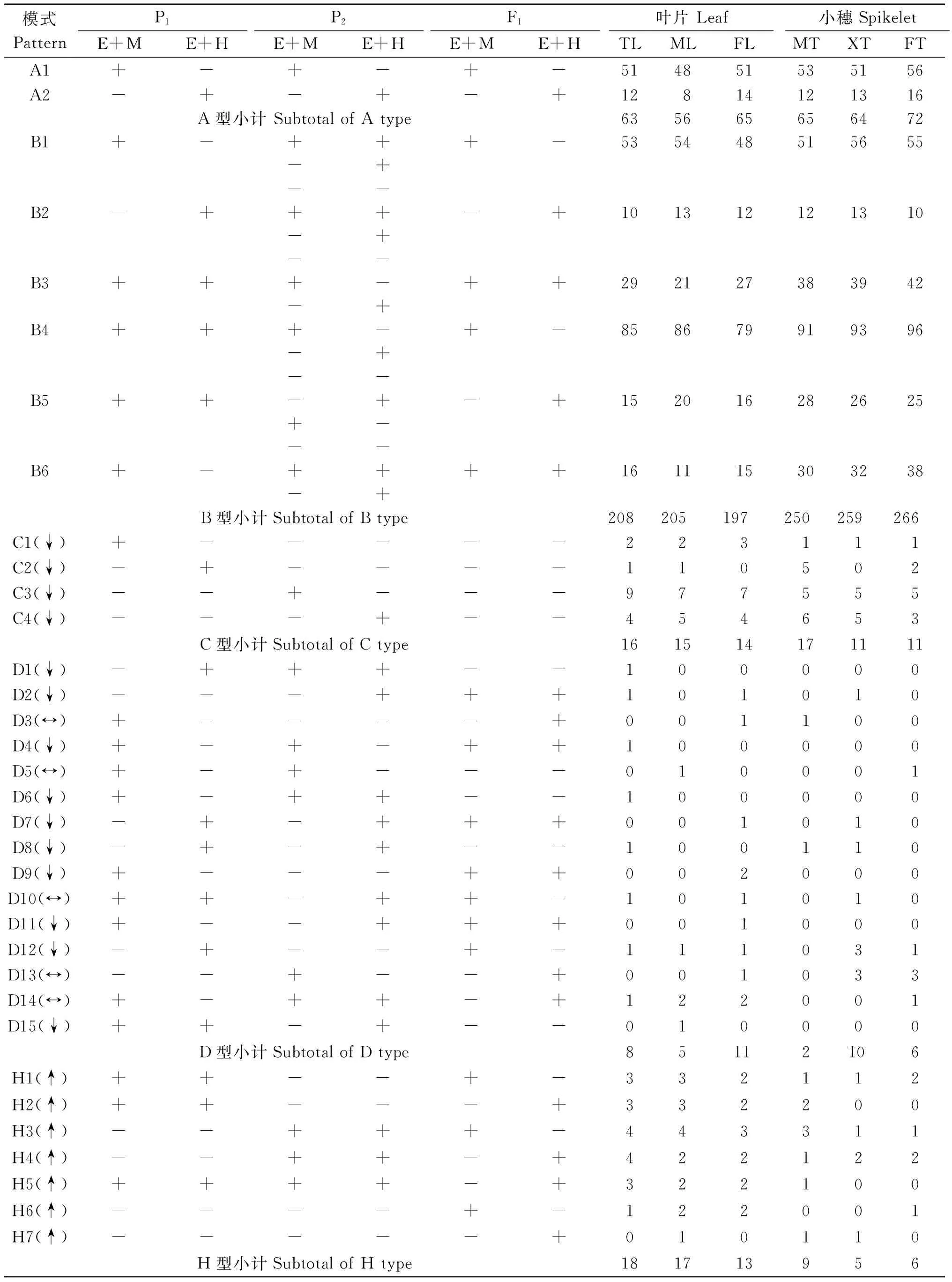
表5 亲本及杂种F1甲基化模式与检测的位点数
注:+.在相应酶切组合中有带;-.在相应酶切组合中无带。
Note:+.There is a band in the enzyme combination;-.No band in the enzyme combination.
由图5可见,在杂种F1DNA甲基化模式的遗传中,A型是杂种与双亲均相同的甲基化模式,即亲本单态型甲基化模式,在叶片和小穗DNA甲基化位点中分别占到20.20%和19.09%。B型、C型和D型的不同亚类分别是遗传自双亲之一的甲基化模式,即双亲多态型甲基化模式,其中遗传自栽培稻亲本辽粳944的亚类有B1、B2、B3、C3和C4,在叶片和小穗DNA甲基化位点中分别占33.26%和32.76%,B3亚类为母本辽粳944与杂种F1均表现为非甲基化的位点,父本则表现为全甲基化或半甲基化,而C3和C4亚类中在父本药用野生稻表现为全甲基化(C3)或半甲基化(C4)的位点,则在母本辽粳944和杂种F1均无CCGG位点,表明杂种F1在B3和C型的状态为去甲基化。此外,杂种F1遗传自父本药用野生稻的甲基化模式有B4、B5、B6、C1和C2亚类,分别在叶片和小穗DNA甲基化位点总数中占到38.64%和44.54%,这表明在亲本甲基化位点向F1的遗传中,杂种F1更多的是遗传了药用野生稻亲本的甲基化位点。
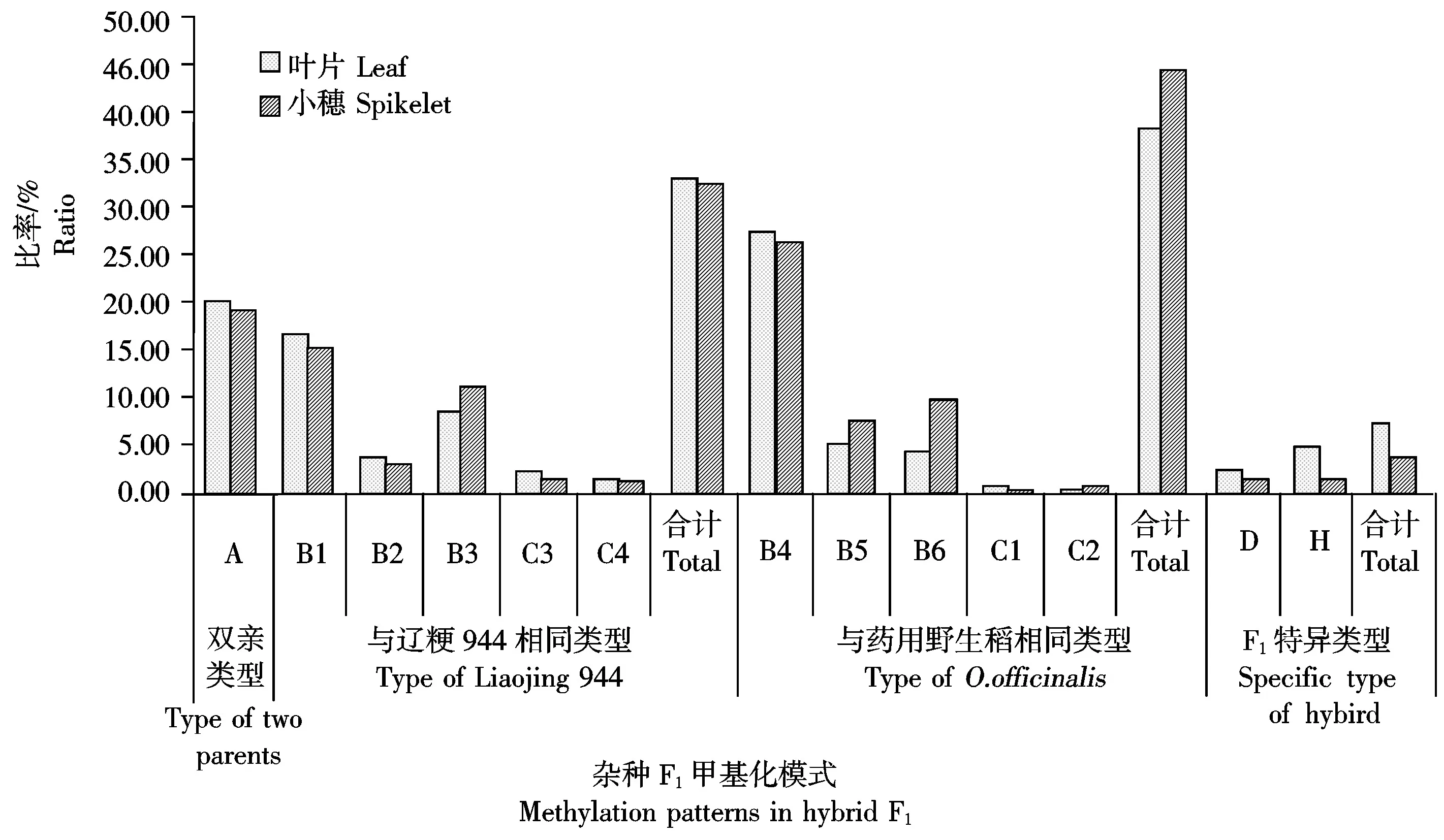
图5 杂种F1甲基化模式遗传与变异的类型及比率
杂种F1DNA甲基化模式的变异指的是在相同酶切位点杂种F1的MSAP带型与2个亲本均不相同,即D型和H型(图5)。杂种F1叶片和小穗DNA甲基化变异的平均概率分别为7.90%和3.61%,其中D型只在极个别发育时期的叶片或小穗DNA中发生,占杂种叶片和小穗DNA总甲基化位点数的2.63%和1.71%,表现为无CCGG位点(表5中的D1、D5、D6、D8和D15)或去甲基化(表5中的D2、D4、D7、D9和D11)或全甲基化(D10和D12)或半甲基化(表5中的D3、D13和D14)等;而H型变异则是杂种F1在双亲非甲基化位点发生了甲基化,为新增加的甲基化位点,其甲基化状态表现为全甲基化或半甲基化,在叶片和小穗组织DNA甲基化位点发生的平均概率分别为5.27%和1.90%。
2.3 甲基化特异片段序列分析与RT-PCR表达分析
从不同发育时期叶片和小穗的MSAP条带中挑选了94个DNA甲基化特异片段用于克隆和测序,并将得到的序列在NCBI网站利用Blast对nr数据库进行同源性比对,结果表明,在94个甲基化特异片段中,有38个片段的序列与已知或假定功能的基因具有同源性(表6),这些基因的功能涉及多个方面,包括植物生长发育、生化代谢及物质转运相关基因(表达的蛋白如类似胚胎发生跨膜蛋白、假定β-葡萄糖苷酶、假定五肽重复包含蛋白质、假定ABC转运蛋白、假定蔗糖磷酸酶等)、信号传导基因(表达的蛋白如锌指蛋白)、抗非生物胁迫及生物胁迫相关基因(表达的蛋白如PHD指转录因子样蛋白、假定热休克蛋白、假定枯草杆菌蛋白酶类丝氨酸蛋白酶、假定蔗糖磷酸酶、假定植物螯合肽合成酶、丝氨酸/苏氨酸蛋白磷酸酶 PP2A-3 催化亚基等)、转座子、反转录转座子或反转座子有关基因(表达的蛋白如假定转座子蛋白、假定反转录转座子蛋白、反转录转座子 p-SINE1-r606等),其中多达11个片段与转座相关的基因具有同源性,表明转座子相关位点是甲基化的热点区域。
根据同源性比对的结果,挑选部分与已知或假定功能的基因具有同源性的甲基化特异片段、以其序列设计引物,并以亲本及杂种相应组织的cDNA为模板,进行甲基化特异片段的RT-PCR表达分析。结果如图6所示,在对21个甲基化特异片段的RT-PCR表达分析中,有6个片段的同源基因表现出了表达的特异性。其中在叶片中,杂种F1在片段FL2的基因表达量介于双亲之间;片段FL12只在母本辽粳944中有表达,在父本和F1中未表达;片段FL14只在父本药用野生稻中表达,在母本和F1中未表达。在小穗中,杂种F1在片段MT5、MT14、XT14的基因表达量相对于亲本则明显上调,涉及的相关同源性基因包括蔗糖磷酸酶、反转录转座子蛋白和锌指蛋白家族基因等。根据这些特异片段的来源情况看,FL2及FL14的甲基化特异片段来自药用野生稻,RT-PCR的基因表达量在材料间趋势不一,而且在FL2和FL14中药用野生稻的基因表达量明显高于栽培稻和杂种F1。尽管MT5、MT14和XT14片段的甲基化发生在杂种F1,亲本在相应位点表现为非甲基化或无CCGG位点,但杂种的基因表达水平却明显高于亲本。推测药用野生稻FL2和FL14位点、杂种F1MT5、MT14和XT14位点的甲基化可能发生在基因的非结构区域或内含子区,或者该位点基因的甲基化与基因表达水平关系不密切。
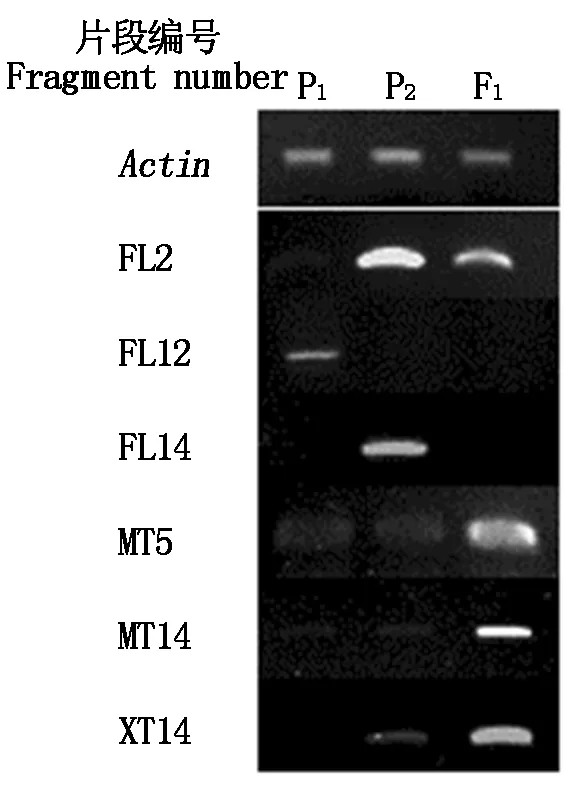
图6 部分甲基化特异片段的RT-PCR结果Fig.6 RT-PCR results of some specific methylated fragments in hybrid F1 and parents
3 讨论
基因组DNA甲基化现象在植物中普遍存在,而且有明显的时空特异性。本研究经过对亚洲栽培稻品种辽粳944(母本)、药用野生稻(父本)及其杂种F1不同发育时期叶片和小穗基因组DNA甲基化遗传与变异特性的系统研究,结果表明辽粳944、药用野生稻及其杂种F1的叶片DNA平均总甲基化水平分别为18.18%,19.76%和20.19%,其小穗DNA平均总甲基化水平分别为16.64%,17.78%和17.38%,叶片DNA甲基化水平略高于小穗;各供试材料叶片和小穗基因组DNA甲基化水平的时空变化趋势相同,即随着生长发育的进行,总甲基化率、全甲基化率和半甲基化率均呈下降趋势,且全甲基化率均显著高于半甲基化率(P=0.000),杂种小穗的半甲基化率显著高于母本辽粳944(P=0.015)。这与前人在拟南芥[22]、水稻[16,18,23]等植物叶片在发育过程的甲基化水平的变化趋势一致。此外,本研究中DNA甲基化水平在叶片和小穗间的组织差异性,在前人研究中也有类似表现,如在玉米中苞叶的甲基化水平最高、雄花穗最低,成熟叶片的甲基化水平高于种子[24];又如高粱的杂种F1和亲本胚乳的甲基化程度均显著低于叶片[25]。推测可能是幼嫩组织处于旺盛的生长过程中,需要启动更多基因的表达参与代谢活动,因此去甲基化程度加大,甲基化水平降低;而相对于叶片等体细胞组织,与生殖有关的组织(小穗、雌雄蕊等)表现较低的甲基化程度也与生殖器官的特殊功能及活跃基因表达的需要有关。
不同植物及不同交配方式产生后代的甲基化遗传变异研究表明,植物DNA甲基化的遗传与变异现象复杂,但具有一定的稳定性,同时在一些杂种中甲基化受到RNA的介导[26]。Takamiya 等[17]利用限制性标记基因组扫描(RLGS)方法、对水稻品种Nipponbare× Kasalath 杂种F1的DNA甲基化遗传性的研究中,亲本的大多数甲基化位点能够遗传给杂种F1,同时存在约4%的异常传递,即亲本甲基化位点在F1丢失即去甲基化或F1产生了新的甲基化位点。在外源染色体片段渗入系的研究中,Dong等[27]发现含有野生稻 (Zizanialatifolia) DNA 片段的栽培稻渗入系在反转录转座子Tos17的5′-LTR区和逆转录酶区都发生了甲基化,包括亲本条带的缺失和新条带的形成,而且这种甲基化变异稳定遗传到了下一代。此外,Nakamura等[28]发现马铃薯自交系(S1~S5代)的甲基化遗传率较高。本研究中杂种F1叶片和小穗DNA的甲基化模式中遗传自双亲的模式类型分别占20.20%和19.09%,遗传自栽培稻亲本的甲基化模式类型分别占33.26%和32.76%,遗传自药用野生稻的占38.64%和44.54%,杂种叶片和小穗DNA在亲本甲基化位点发生变异的比率分别为7.90%和3.61%,主要表现为超甲基化,且半甲基化率显著提高。可见,杂种叶片和小穗甲基化模式以遗传为主,而且更多的是遗传了药用野生稻的甲基化位点。Jin等[19]的研究表明,栽培稻与药用野生稻种间杂种F1开花期旗叶DNA的甲基化变异率接近7.6%,其中F190.9% 的甲基化位点变异在BC1F1得到保持。这与本研究中杂种叶片3个发育时期平均甲基化率变异率(7.90%)非常接近,可以认为栽培稻和药用野生稻的基因组DNA甲基化具有相对较高的稳定性,也证明了MSAP技术用于研究植物DNA甲基化的可靠性。
尽管植物DNA甲基化特异性与功能基因的关系以及对基因表达的影响是表观遗传研究中重要的研究领域之一,但目前在不同植物的研究中,主要是通过间接推测,认为基因甲基化与表型变异之间有密切关系。Akimoto等[29]则提供了直接证据,证明基因去甲基化后产生了表达效应,形成了表型。他们利用氮杂脱氧胞苷处理水稻种子以去甲基,经连续10年对处理种子后代进行种植,并接种水稻白叶枯病菌观察发病情况及检测基因组甲基化状态,发现其中1个株系Line-2表现了对白叶枯病的抗性,同时与野生型比较,株系Line-2的Xa21G基因启动子发生了去甲基化,从而证明该株系的水稻白叶枯病抗性是Xa21G基因表达的结果。Zhang等[30]对不同倍性水稻的研究表明,基因启动子区甲基化与基因表达有明确的关系,但基因结构区甲基化与基因表达的负相关关系不明显。Li等[31]研究表明,基因超甲基化并不一定使其基因表达水平下降。在本研究中,杂种F1甲基化特异片段MT5、MT14和XT14的RT-PCR基因表达量在3个材料间趋势不一致,尽管MT5、MT14和XT14的甲基化发生在杂种F1,亲本在相应位点表现为非甲基化或无CCGG位点,但是这些位点杂种的基因表达水平却明显高于亲本。综合来看,基因甲基化可发生在基因特异位点,从而表现出与基因表达的复杂关系,需要开展不同物种及基因甲基化特点的更深入研究。
本研究对杂种及亲本叶片和小穗的94个甲基化特异片段经测序和同源性比对分析,有38个片段的序列与已知或假定功能的基因具有同源性,这些基因的功能种类涉及多个方面,包括植物生长发育、生化代谢及物质转运相关基因、信号传导基因、抗非生物胁迫及生物胁迫相关基因、转座子等,其中多达11个片段与转座相关的基因具有同源性。但在小穗的甲基化特异片段序列分析中,未发现与育性密切相关的特异基因,可能本研究中的种间杂种F1败育受基因组甲基化的影响较小。尽管前人在烟草、小麦、玉米等[11-12]的研究中,对花粉粒、或雄性不育系、或雄性可育杂种和雄性不育杂种的甲基化研究表明,与生殖发育及育性有关组织的DNA甲基化有一定的特异性,而且在一定程度上与基因的表达调节变化有关,但也未能明确具体的调控基因及其机理。因此,今后需要进一步开展包括水稻远缘杂交及杂种败育的表观遗传规律的研究,而且在深入开展基因组DNA甲基化研究的同时,有必要开展非编码RNA调控和染色质重塑等表观遗传机制的研究,这不仅有助于深入了解远缘杂交引起的基因组冲突与表观遗传的关系,更有助于多方位了解远缘杂种不育的形成机制,对探索克服远缘杂种不育的有效途径和野生种质资源的高效利用都具有十分重要的意义。
[1] Brar D S,Khush G S. Alien introgression in rice[J]. Plant Molecular Biology,1997,35(1/2):35-47.
[2] 何光存. 野生稻有利基因的挖掘与转移[M].//罗利军,应存山,汤圣祥.稻种资源学. 武汉:湖北科学技术出版社,2002:279.
[3] Jena K K,Khush G S. Introgression of genes fromOryzaofficinalisWell ex Watt to cultivated rice,O.sativaL.[J]. Theoretical and Applied Genetics,1990,80(6):737-745.
[4] Huang Z,He G,Shu L,et al. Identification and mapping of two brown planthopper resistance genes in rice[J]. Theoretical and Applied Genetics,2001,102(6/7):929-934.
[5] Fu X L,Lu Y G,Liu X D,et al. Cytological behavior of hybridization barriers betweenOryzasativaandOryzaofficinalis[J]. Agricultural Sciences in China,2011,10(10):1489-1500.
[6] Li G,Tang M,Hu W,et al. Characterization of an Alien chromosome ofOryzaofficinalistransferred the genomic and cytological environment ofOryzasativa[J]. Journal of Plant Biology,2010,53(4):306-313.
[7] Mcclintock B. The significance of responses of the genome to challenge[J]. Science,1984,226(4676):792-801.
[8] Dong Z Y,Wang Y M,Zhang Z J,et al. Extent and pattern of DNA methylation alteration in rice lines derived from introgressive hybridization of rice andZizanialatifoliaGriseb[J]. Theoretical and Applied Genetics,2006,113(2):196-205.
[9] Ran L P,Fang T T,Rong H,et al. Analysis of cytosine methylation in early generations of resynthesizedBrassicanapus[J]. Journal of Integrative Agriculture,2016,15(6):1228-1238.
[10] Baroux C,Raissig M T,Grossniklaus U. Epigenetic regulation and reprogramming during gamete formation in plants[J]. Current Opinion in Genetics & Development,2011,21(2):124-133.
[11] Ba Q S,Zhang G S,Niu N,et al. Cytoplasmic effects on DNA methylation between male sterile lines and the maintainer in wheat (TriticumaestivumL.) [J]. Gene,2014,549(1):192-197.
[12] Chen B,Zhang Y,Lu Y L,et al. DNA methylation analysis of sterile and fertile CMS-C hybrids and their parents in maize[J]. Journal of Plant Biochemistry and Biotechnology,2016,25(1):3-11.
[13] Wang W,Huang F,Qin Q,et al. Comparative analysis of DNA methylation changes in two rice genotypes under salt stress and subsequent recovery[J]. Biochemical and Biophysical Research Communications,2015,465(4):790-796.
[14] Komiya R,Ikegami A,Tamaki S,et al. Hd3a and RFT1 are essential for flowering in rice[J]. Development,2008,135(4):767-774.
[15] Fujino K,Sekiguchi H. Site specific cytosine methylation in rice nonautonomous transposable element nDart[J]. Plant Molecular Biology,2008,67(5):511-518.
[16] Xiong L Z,Xu C G,Saghai Maroof M A,et al. Patterns of cytosine methylation in an elite rice hybrid and its parental lines,detected by a methylation-sensitive amplification polymorphism technique[J]. Molecular & General Genetics,1999,261(3):439-446.
[17] Takamiya T,Hosobuchi S,Noguchi T,et al. Inheritance and alteration of genome methylation in F1hybrid rice[J]. Electrophoresis,2008,29(19):4088-4095.
[18] Sakthivel K,Girishkumar K,Ramkumar G,et al. Alterations in inheritance pattern and level of cytosine DNA methylation,and their relationship with heterosis in rice[J]. Euphytica,2010,175(3):303-314.
[19] Jin H,Hu W,Wei Z,et al. Alterations in cytosine methylation and species-specific transcription induced by interspecific hybridization between OryzasativaandO.officinalis[J]. Theoretical and Applied Genetics,2008,117(8):1271-1279.
[20] Murray M G,Thompson W F. Rapid isolation of high molecular weight plant DNA[J]. Nucleic Acids Research,1980,8(19):4321-4325.
[21] Reyna-López G E,Simpson J,Ruiz-Herrera J. Differences in DNA methylation patterns are detectable during the dimorphic transition of fungi by amplification of restriction polymorphisms[J]. Molecular & General Genetics ,1997,253(6):703-710.
[22] Finnegan E J,Peacock W J,Dennis E S. Reduced DNA methylation inArabidopsisthalianaresults in abnormal plant development[J]. Proceedings of the National Academy of Sciences of the United States of America,1996,93(16):8449-8454.
[23] 潘雅姣,傅彬英,王 迪,等. 水稻干旱胁迫诱导DNA甲基化时空变化特征分析[J]. 中国农业科学,2009,42(9):3009-3018.
[24] Lu Y,Rong T,Cao M. Analysis of DNA methylation in different maize tissues[J]. Journal of Genetics and Genomics,2008,35(1):41-48.
[25] Zhang M S,Yan H Y,Zhao N,et al. Endosperm-specific hypomethylation,and meiotic inheritance and variation of DNA methylation level and pattern in sorghum (SorghumbicolorL.) inter-strain hybrids[J]. Theoretical and Applied Genetics,2007,115(2):195-207.
[26] Zhang Q,Wang D,Lang Z,et al. Methylation interactions inArabidopsishybrids require RNA-directed DNA methylation and are influenced by genetic variation[J]. Proceedings of the National Academy of Sciences of the United States of America,2016,113(29):E4248-E4256.
[27] Dong Y Z,Liu Z L,Dong Y S,et al. Alien DNA introgression into rice causes heritable alterations in DNA methylation patterns in an active retrotransposon Tos17[J]. Acta Botanica Sinica,2004,46(1):100-109.
[28] Nakamura S,Hosaka K. DNA methylation in diploid inbred lines of potatoes and its possible role in the regulation of heterosis[J]. Theoretical and Applied Genetics,2010,120(2):205-214.
[29] Akimoto K,Katakami H,Kim H J,et al.Epigenetic inheritance in rice plants[J].Annals of Botuny,2007,100(2):205-217.
[30] Zhang H Y,Zhao H X,Wu S H,et al. Global methylation patterns and their relationship with gene expression and small RNA in rice lines with different ploidy[J]. Frontiers in Plant Science,2016,7:1002.
[31] Li S,Zhu Y,Zhi L,et al. DNA methylation variation trends during the embryonic development of chicken[J]. PLoS One,2016,11(7):e0159230.
Inheritance and Variations of DNA Methylation in Interspecific Hybrid F1BetweenO.sativaandO.officinalis
HE Ping1,SHU Mian1,CAI Xiaodan1,FU Xuelin2
(1.College of Life Sciences,South China Agricultural University,Guangzhou 510642,China;2. College of Agriculture,South China Agricultural University,Guangzhou 510642,China)
Aiming to analysis the characteristics of DNA methylation during the development processes of leaf and spikelet to explore the effect of genome shock in different tissues of hybrid F1betweenO.sativaandO.officinalis,moreover,the possibility of biological effects,in this study,using the methylation sensitive amplified polymorphism technology (MSAP),patterns and levels of global genomic DNA methylation in leaf and spikelet from parents to interspecific hybrid F1were investigated. The results showed that,the similar change trends of DNA methylation level in tissues of leaf and spikelet were observed in all the experimental materials,that is,along with the plant growth and development the DNA methylation level was on a downward trend,and the full methylation level was significantly higher than hemi-methylation level in both leaf and spikelet(P=0.000). In hybrid leaf tissue at the tillering stage, meiosis stage of pollen mother cells and flowering stage,the average total methylation level,full methylation level and hemi-methylation level was 20.19%,16.06% and 4.13%,respectively,which was higher than that of the two parents;while in hybrid spikelet tissue at the meiosis stage of pollen mother cells, microspore development stage and matured pollen grain stage,the average level of total,full and hemi-methylation was 17.38%,13.67% and 3.71%,respectively,which was higher than that of Liaojing 944 and lower thanO.officinalis. However,only hemi-methylation level of hybrid was significantly higher than Liaojing 944(P=0.015). DNA methylation patterns in leaf and spikelet were very similar in parents and hybrid. Sequence analysis of 94 differentially methylated fragments showed that 38 fragments were sequence similarity in the NCBI database. The results would be helpful for further analysis of hybrid sterility betweenO.sativaandO.officinalis.
Oryzasativa;Oryzaofficinalis;Interspecific hybrid;DNA methylation;Methylation sensitive amplified polymorphism (MSAP)
2017-06-20
广东省自然科学基金项目(9151063201000057);广东省教育厅“创新强校工程”自主创新能力提升类项目(2014KTSCX032);广东省科技厅省级科技计划项目(2015A030302067);公益性行业(农业)科研专项子项目(201003021);国家自然科学基金项目(31671762)
何 平(1967-),男,安徽枞阳人,副教授,博士,主要从事生物化学与分子生物学研究。
傅雪琳(1967-),女,陕西扶风人,教授,博士,主要从事野生稻有利基因发掘与利用研究。
S511.03
A
1000-7091(2017)04-0019-13
10.7668/hbnxb.2017.04.004
