单层GaSe表面Fe原子吸附体系电子自旋性质调控∗
2017-09-07卢奕宏柯聪明付明明吴志明康俊勇张纯淼吴雅苹
卢奕宏 柯聪明 付明明 吴志明 康俊勇 张纯淼 吴雅苹
(厦门大学物理系,福建省半导体材料及应用重点实验室,半导体光电材料及其高效转换器件协同创新中心,厦门 361005)
单层GaSe表面Fe原子吸附体系电子自旋性质调控∗
卢奕宏 柯聪明 付明明 吴志明 康俊勇 张纯淼†吴雅苹‡
(厦门大学物理系,福建省半导体材料及应用重点实验室,半导体光电材料及其高效转换器件协同创新中心,厦门 361005)
(2017年3月7日收到;2017年6月3日收到修改稿)
III族金属单硫化物因其优越的光电和自旋电子特性而备受关注,实现对其自旋性质的有效调控是发展器件应用的关键.本文采用密度泛函理论系统地研究了GaSe表面Fe原子吸附体系的几何构型及自旋电子特性.Fe/GaSe体系中Fe吸附原子与最近邻Ga,Se原子存在较强的轨道耦合效应,使体系呈现100%自旋极化的半金属性.其自旋极化贡献主要来源于Fe-3d电子的转移及Fe-3d,Se-4p和Ga-4p轨道杂化效应.对于Fe双原子吸附体系,两Fe原子之间的自旋局域导致原本从Fe转移至GaSe的自旋极化电荷量减少,从而费米能级附近的单自旋通道转变为双自旋通道,费米能级处的自旋极化率转变为0.研究结果揭示了Fen/GaSe吸附体系自旋极化特性的形成和转变机制,可为未来二维自旋纳米器件的设计与构建提供参考.
表面吸附,密度泛函理论,半金属性,自旋特性调控
1 引 言
石墨烯以其独特的二维结构和丰富优良的物理性质引起了全世界研究者的广泛关注[1].然而,固有的带隙缺失从本质上限制了其在传统发光器件领域的应用[2].于是,科学家们在尝试通过外加电场、掺杂等方式打开石墨烯带隙的同时,也逐渐探索与其相似且本身具有不同带隙宽度的二维材料,以满足器件应用的多重需求[3−5].近年来,III族金属单硫化物MX(M=Ga,In;X=S,Se,Te)作为一种新的层状半导体材料具有优越的电学、光学、磁学等性质,赢得了越来越多科学工作者的青睐[6,7].机械剥离的少层GaSe展现良好光响应性能,在紫外254 nm光照射下其光响应度可达2.8 A/W,相应的量子效率和开关比分别可达1367%和103,优于MoS2和石墨烯等[6,8,9].除了在电子器件和光电器件方面的应用以外,III族金属单硫化物二维材料还具备优异的自旋电子特性.其独特的晶体场各向异性和自旋-轨道耦合作用使轨道电子态的简并消退,在非共振偏振激发下产生具有高度自旋极化的载流子,其自旋极化率可超过90%,且具有较长的自旋弛豫时间[10,11];同时,自旋极化特性可在体材料到纳米单层厚度范围内得以精细调控.由此可见,III-VI族半导体材料同时兼具二维结构、优异的光电性能及独特的自旋电子特性,对于超高密度集成和超快速度运转的自旋光电器件具有重要的潜在应用价值.
为了进一步满足器件的设计需要,还可通过外加应力[12]、调节组分[12]或电荷掺杂[13]等方法实现对自旋磁性的有效调性.目前,人们已通过引入掺杂原子及空位缺陷的方式在GaSe单层材料中产生高自旋极化并实现交互巡回的铁磁性和半金属性[14,15].相形之下,利用吸附原子的表面修饰作用调控单层GaSe电子及自旋磁性的研究却鲜有报道.同时,铁磁材料,如Fe,Co和Ni具有高电子自旋极化率、高居里温度及易于制备等特点[16−18],因此,采用表面吸附方式,在GaSe等III族金属单硫化物二维表面修饰铁磁原子可简便而有效地实现对其电子及自旋磁性的调控.
本文通过第一性密度泛函理论,系统研究了磁性金属Fen(n=1,2)原子对二维单层GaSe自旋电子性质的调控.首先通过吸附能计算确定其最稳定的吸附位,据此计算其自旋分辨能带结构、电子态密度及差分电荷密度以分析Fen/GaSe吸附体系的自旋和电子轨道耦合作用.结果表明,Fe/GaSe体系在费米能级处呈现100%自旋极化的半金属性,而Fe2/GaSe由于两Fe原子之间的自旋局域,体系在费米能级处的自旋极化率转变为0%.
2 理论计算方法
本文采用第一性原理模拟方法,运用基于密度泛函理论的VASP[19]程序包,分别模拟清洁GaSe及Fen吸附的单层GaSe的几何及电子结构.为了隔绝周期性扩展的吸附原子间的相互影响以及材料层间的相互作用,体系采用3×3超原胞(18个Se原子和18个Ga原子)结构并在z方向引入厚度约25Å的真空层.Ga,Se及Fe原子的价电子结构分别为4s24p1,4s24p4和3d64s2.计算采用Perdew-Burke-Ernzerhof[20]交换关联泛函及广义梯度近似(GGA),布里渊区内的积分采用9×9×1的Monkhorst-Pac k点阵,平面波基组的截断能量Ecut为350 eV.结构优化过程中,电子迭代收敛的总能允差设为10−6eV,离子弛豫运动的收敛标准取为10−2eV/Å,以保证晶胞所有原子自由度充分弛豫,体系能量达到最低.
3 结果与讨论
单层GaSe 3×3超原胞几何构型如图1(a)所示.清洁GaSe二维表面呈现六角蜂窝状结构,其优化后的晶格常数为3.82Å[21].Ga—Ga和Ga—Se键长分别为2.47Å和2.50Å,相邻Se原子之间Se—Ga—Se夹角约为99.5◦.鉴于六角蜂窝状结构的几何对称性,GaSe表面存在三个高对称吸附位,分别为芯位H,Se原子顶位T1以及Ga原子顶位T4.Fe原子的吸附能定义为E ad=E GaSe+n×E Fe−E Fen/G aSe,其中E GaSe,EFe和EFen/GaSe分别代表清洁GaSe、孤立Fe原子和Fen/GaSe体系总能,n为吸附的Fe原子个数.计算结果显示Fe单原子于H位、T1位和T4位的吸附能分别为1.08,0.59和2.20 eV,表明Ga原子顶位T4位为Fe原子的最稳定吸附位.由结构模型的原子坐标可知,Fe原子吸附于H位和T1位时,最近邻Ga原子与Se原子的位置基本没有变化,而Fe原子吸附于T4位时,其位置则发生剧烈变化.除此之外,Fe原子吸附于T4位时,Fe—Ga键长及Fe—Se键长分别为2.14Å和2.38Å,比其他两个高对称位的相应键长都要短一些.较短键长通常意味着更大的波函数交叠与更强的原子间相互作用,表明Fe原子吸附T4位时与其最近邻原子间的相互作用比吸附H位和T1位时更强.在获得最稳定Fe/GaSe吸附体系的基础上,我们进一步研究吸附第二个Fe原子的Fe2/GaSe体系的电子性质(两个Fe原子分别标记为Fea和Feb).考虑结构的对称性,Feb的可能吸附位选择为Fea的顶位T位以及Fea的最近邻T1位、H位和T4位,弛豫后的结构模型和各主要原子间键长如图1(e)—(h)及表1所列.Feb吸附T位时,Feb只与Fea存在直接相互作用,Feb—Fea键长为2.17Å;下方GaSe受Feb影响较小,体系中其他原子的相对位置与Fe/GaSe体系基本一致,如图1(e)所示.Feb吸附T1位时,由于与Fea的成键相互作用而彼此靠近,Feb—Fea键长为2.13Å;同时Fea正下方的Ga原子也移至两Fe原子之间并与之成键.当Feb吸附H位时,最近邻Ga原子由于H位Fe原子的作用而偏离原本位置并向其靠近,此时Feb—Fea键长为2.29Å,Fea—Ga和Feb—Ga键长几乎相等.而当Feb吸附T4位时,由于Fe原子之间的相互作用,经弛豫后Fea原子移至H位附近.可见,Feb吸附T4位时体系处于非稳定状态,相比之下,其余三种构型的Fe2/GaSe体系为稳定或亚稳态.计算结果表明,Feb原子四个吸附位置的吸附能分别为1.67,1.99,3.67和3.58 eV,因此H位为Feb的最稳定吸附位.因为Feb原子吸附于H位和T4位的弛豫结构基本一致,故两者吸附能也较为相近.

图1 (网刊彩色)弛豫后(a)清洁的单层GaSe、吸附Fe单原子于GaSe(b)H位、(c)T1位及(d)T4位及Feb吸附于Fe/GaSe的(e)T位、(f)T1位、(g)H位及(h)T4位的结构模型俯视图Fig.1.(color on line)Top vieWs of(a)the relaxed GaSe ML,of the relaxed Fe/GaSe systeMWith a Fe adatoMlocating on H(b),T1(c),and T4(d)sites of GaSe su rface,and of the relaxed Fe2/GaSe systeMWith a FebadatoMlocating on T(e),T1(f),H(g)and T4(h)site of Fe/GaSe surface,respectively.

表1 Fen/GaSe系统结构参数:不同吸附位Fe原子之间键长及其与GaSe最近邻Ga,Se原子间键长,表中所有的长度单位均为ÅTab le 1.Structu ral paraMeters of Fen/GaSe system:bond lengths of Fe adatoMs and their nearest neighbor atoMs on their diff erent adsorp tion sites.A ll lengths are inÅ.
为了阐明Fen/GaSe体系的自旋电子特性,我们计算了其自旋分辨的能带结构.清洁单层GaSe自旋向上与自旋向下能带结构完全重合,表明其具有非磁性基态,如图2(a)所示.其导带底位于Γ点,价带顶位于Γ点与M点之间,使之呈现间接能隙结构,其值为1.83 eV,与之前文献结果一致[22].吸附一个Fe原子后,对称分布的能带分裂为两个非对称的自旋向上和向下的通道,其中价带顶由原来的位置移至Γ点,体系随即转变为直接带隙,如图2(b)和图2(c)所示.费米能级附近出现两条杂质能级,并仅对自旋向下通道有贡献.同时由于Fe与周边原子的轨道杂化,部分电荷由Fe-4s轨道转移至3d轨道,从而导致约2个不成对电子,产生约为2.122µB的磁矩,并在最近邻Se和Ga原子中分别形成约−0.004µB和−0.178µB的诱导磁矩(表2),意味着体系中Fe吸附原子与其周围原子间存在反铁磁耦合效应.为了验证磁矩的稳定性,我们计算了体系的自旋极化能,其定义为体系电子有无自旋极化的能量差.计算结果表明其自旋极化能为0.02 eV/atom,足够保持磁矩的稳定性.随着第二个Fe原子的吸附,原本Fe/GaSe体系自旋向上通道的价带顶能级向上移动至费米能级附近,导带底与费米级之间也出现一条新的杂质能级,而自旋向下通道的杂质能级在导带底和价带顶附近也均有明显增加.然而,与Fe单原子吸附的Fe/GaSe不同的是,Fe2/GaSe体系两个自旋通道在费米能级处均无能级分布.两Fe原子磁矩分别为2.451µB和1.690µB,表明存在铁磁耦合效应;最近邻Se原子及Ga原子的诱导磁矩分别为0.031µB和−0.181µB,表明Feb原子吸附后两Fe原子与附近Ga原子仍存在反铁磁耦合效应,但与近邻Se原子的耦合效应则由原本的反铁磁耦合转变为铁磁耦合.此外,我们还计算了Fe2/GaSe体系中两Fe原子之间铁磁耦合的稳定性,计算结果显示Fe原子之间的铁磁耦合较反铁磁耦合状态更为稳定,其能量低差约为0.33 eV.

图2 (网刊彩色)(a)完整GaSe单层、(b),(c)Fe/GaSe及(d),(e)Fe2/GaSe体系自旋分辨的能带结构Fig.2.(color on line)(a)Energy band structu res of(a)the p ristine GaSe ML,(b),(c)Fe/GaSe,and(d),(e)Fe2/GaSe systeMs.

表2 Fen/GaSe体系中Fe原子及其最近邻Se和Ga原子的电荷转移和磁矩Tab le 2.Diff erential charge and MagneticMoMents of the Fe adatoMs and their nearest neighbor Se and Ga atoMs of Fen/GaSe systeMs.
自旋分辨的总态密度(TDOS)如图3所示.清洁的单层GaSe总态密度呈现上下对称分布,与能带结构计算结果一致.相比之下,Fe/GaSe的费米能级向高能方向移动,且带隙减小约0.55 eV,同时费米能级附近仅出现自旋向下的电子态,且费米能级处自旋极化率为100%,表现为单自旋的半金属性[23].随着第二个Fe原子的吸附,Fe2/GaSe体系的费米能级继续向高能方向移动,同时带隙进一步减小.原先位于费米能级附近的单自旋通道转变为双自旋通道同时存在,且由于能级的移动,费米能级处的自旋极化率由原来的100%转变为0.进而,我们通过对Fen/GaSe吸附体系中Fe原子及其相邻Se和Ga原子分波态密度(PDOS)的分析,深入了解自旋极化态的轨道贡献,如图4所示.Fe/GaSe体系中Fe吸附原子的电子态主要占据导带底0.75—0.86 eV能量区间的自旋向下通道以及价带顶−0.83—−0.55 eV能量区间的自旋向上通道,其贡献主要源自于Fe原子3d态.对于Fe最近邻的Se原子,4p态在价带顶−0.83—−0.55 eV的自旋向上通道中有明显占据,而4s态的作用相对比较小.不同于Se原子,最近邻Ga原子的电子态在价带顶贡献较小,其4s态和4p态主要占据导带底附近的自旋向上通道.除了占据导带底和价带顶的耦合态,Fe原子及其最近邻的Ga和Se原子亦在费米能级附近出现一些自旋极化的杂质能级,它们只占据自旋向下通道,导致出现半金属性传导特性.而对于Fe2/GaSe体系,两个Fe吸附原子的电子态主要占据导带底0.35—0.43 eV能量区间的上下自旋通道、价带顶−0.69—−0.55 eV能量区间的自旋向下通道和带间−0.32—−0.09 eV能量区间的上下自旋通道,贡献主要来源于Fe原子3d态.对于两个Fe最近邻的Se原子,4p态在导带顶0.35—0.43 eV的自旋向上通道和带间−0.32—−0.09 eV能量区间的两个自旋通道中均有明显占据,而4s态的贡献相对较小.最近邻Ga原子的电子态在带间贡献比较小,其4s态和4p态主要占据导带底0.35—0.43 eV能量区间的自旋向上通道和价带顶−0.69—−0.55eV能量区间的自旋向下通道.由图4可知,两吸附体系中无论是在费米能级附近的价带顶还是导带底,Se-4p,Ga-4p和4s轨道的分波态与两个Fe吸附原子的3d轨道的分波态均十分符合,表明两个Fe原子之间、Fe原子与其最近邻原子的波函数高度交叠,可知Fe与GaSe存在明显的成键相互作用.

图3 (网刊彩色)(a)清洁GaSe单层、(b)Fe/GaSe和(c)Fe2/GaSe吸附体系的总态密度图Fig.3.(color on line)TDOS of(a)the p ristine GaSe ML,(b)Fe/GaSe and(c)Fe2/GaSe adsorp tion systeMs.
为了澄清Fen/GaSe吸附体系自旋极化特性的形成及转变机制,进一步模拟Fe吸附原子与GaSe单层之间的相互作用及电荷分布.体系差分空间电荷密度∆ρ(r)可通过以下公式计算:


图4 (网刊彩色)Fe/GaSe吸附体系中Fe原子(a)与其最近邻Se原子(b)和Ga原子(c)及Fe2/GaSe体系中两个Fe原子(d),(e)与其最近邻的Se原子(f)和Ga原子(g)的分波态密度,实线和虚线分别代表向上和向下自旋电子态Fig.4.(color on line)PDOS of the Fe adatom(a),its nearest neighbor Se(b)and Ga(c)atoMs of Fe/GaSe,and Fe adatoMs(d),(e),their nearest neighbor Se(f)and Ga(g)atoMs of Fe2/GaSe.The solid and dashed lines denote theMa jority and Minority spin states,respectively.
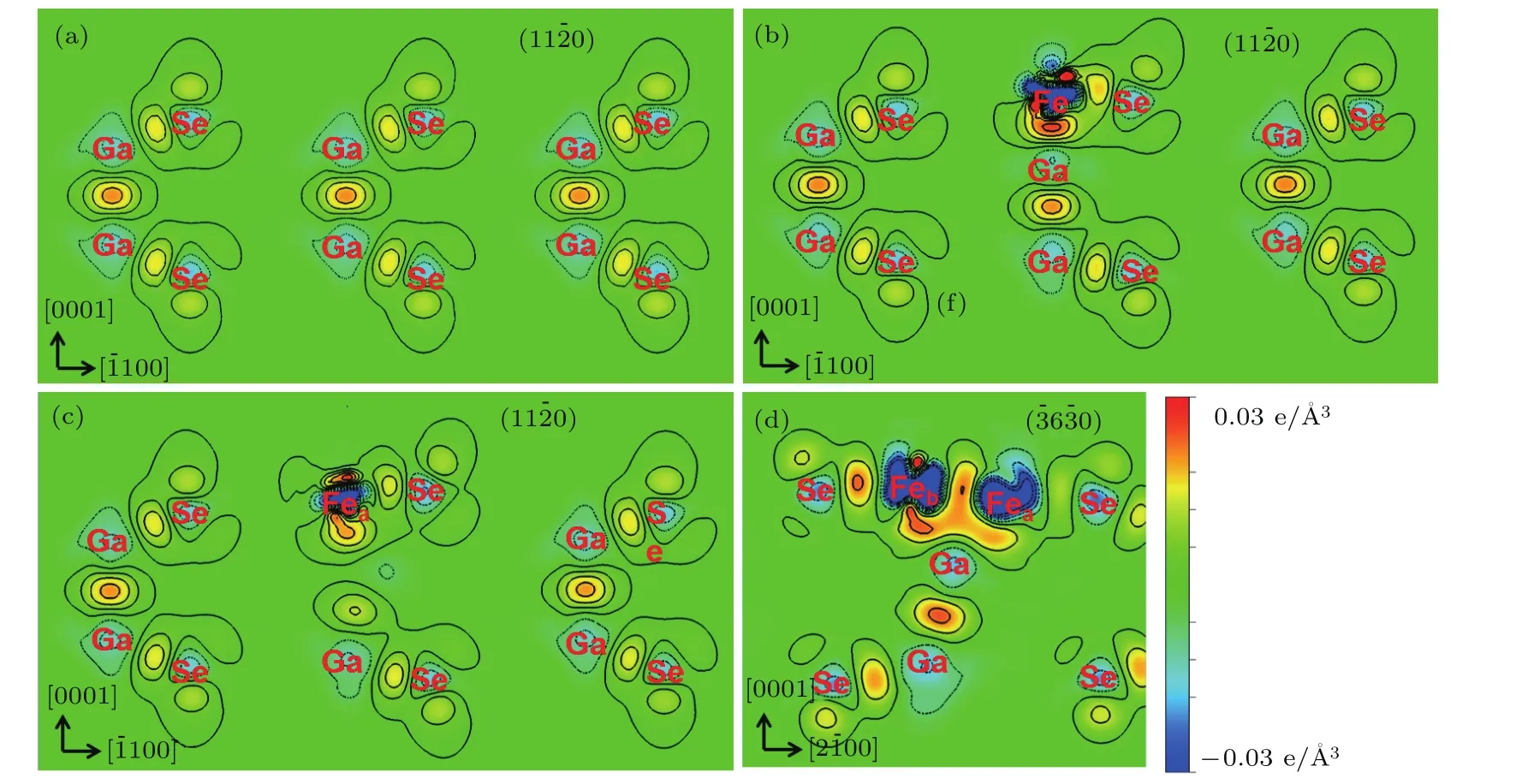
图5 (网刊彩色)(a)未吸附Fe原子、(b)吸附一个Fe原子及(c),(d)吸附两个Fe原子的单层GaSe模型(110)和(60)面的差分面电荷密度,其中负值和正值的等值线分别用蓝色虚线和黑色实线表示,相邻等值线的间距为0.003 e.Å−3Fig.5.(color on line)The vertical sections of deforMation charge densities of the(110)p lane of GaSe ML(a)Withou t Fe atoMadsorp tion,(b)With one adsorbed Fe atom,(c)and(d)With two adsorbed Fe atoMs.The negative and positive contours are respectively represented by the blue dashed lines and b lack solid lines With the contour interval of 0.003 e.Å−3.
4 结 论
本文通过第一性原理密度泛函理论,系统地研究了GaSe表面Fe原子吸附体系的几何结构及自旋电子特性.单层GaSe表面,Ga原子顶位T4为Fe单原子最稳定吸附位,而T4-H位为Fe双原子最稳定吸附位.Fe单原子吸附的Fe/GaSe体系中Fe原子与最近邻Ga,Se原子之间存在很强的反铁磁耦合效应,使体系呈现100%自旋极化的半金属性,其自旋极化贡献主要来源于Fe-3d电子的转移及Fe-3d,Se-4p和Ga-4p轨道杂化.对于Fe双原子吸附体系,两Fe原子之间的强相互作用导致原本从Fe转移至GaSe的自旋极化电荷量的减少,从而费米能级附近的单自旋通道也随之转变为双自旋通道,Fe原子与最近邻Se原子的耦合效应也由原本的反铁磁耦合转变为铁磁耦合;同时由于能级的移动,费米能级处的自旋极化率转变为0,本文揭示了Fen/GaSe吸附体系自旋极化特性的形成和转变机制,对未来二维自旋纳米器件的设计与构建具有一定的参考意义.
[1]Novoselov K S,GeiMA K,Morozov SV,Jiang D,Zhang Y,Dubonos S V,Grigorieva IV,Firsov A A 2004 Science 306 666
[2]Zhou SY,Gweon G H,Fedorov A V,First P N,de Heer WA,Lee D H,Guinea F,Neto A H C,Lanzara A 2007 Nat.Mater.6 770
[3]Song L,Ci L J,Lu H,Sorokin P B,Jin C H,Kvashnin A G,Kvashnin D G,Lou J,Yakobson B I,A jayan P M2010 Nano Lett.10 3209
[4]Bianco E,Butler S,Jiang SS,Restrepo O D,Wind lW,Goldberger J E 2013 ACS Nano 7 4414
[5]Wu S F,Buck ley S,Schaib ley J R,Feng L F,Yan J Q,Mand rus D G,HataMi F,Yao W,Vuckovic J,Ma juMdar A,Xu X D 2015 Nature 520 69
[6]Late D J,Liu B,Luo J J,Yan A M,Matte H S S R,G rayson M,Rao C N R,D ravid V P 2012 Adv.Mater.24 3549
[7]Hu P,Wang L,Yoon M,Zhang J,Feng W,Wang X,Wen Z,Id robo J C,MiyaMoto Y,Geohegan D B,X iao K 2013 Nano Lett.13 1649
[8]Late D J,Liu B,Matte H S S R,Rao C N R,D ravid V P 2012 Adv.Fun.Mater.22 1894
[9]Hu P A,Wen Z Z,Wang L F,Tan P H,X iao K 2012 ACS Nano 6 5988
[10]GaMarts E M,Ivchenko E L,KaraMan MI,Mushinski V P,Pikus G E,Razbirin B S,Starukhin A N 1977 Sov.Phys.JETP 46 590
[11]Ivchenko E L,Pikus G E,Razbirin B S,Starukhin A I 1977 Sov.Phys.JETP 45 1172
[12]WeiW,Dai Y,Liu C W,Ma Y D,Huang B B 2015 J.Mater.Chem.C 3 11548
[13]Cao T,Li Z L,Louie S G 2015 Phys.Rev.Lett.114 236602
[14]Peng Y T,X ia C X,Zhang H,Wang T X,Wei S Y,Jia J 2014 Phys.Chem.Chem.Phys.16 18799
[15]Ao L,X iao H Y,X iang X,Li S,Liu K Z,Huang H,Zu X T 2015 Phys.Chem.Chem.Phys.17 10737
[16]Wang WG,Li M,HageMan S,Chien C L 2012 Nat.Mater.11 64
[17]Ikeda S,Miura K,YaMaMoto H,MizunuMa K,Gan H D,Endo M,K anai S,Hayakawa J,Matsukura F,Ohno H 2010 Nature Mater.9 721
[18]MaruyaMAt,Shiota Y,Nozaki T,Ohta K,Toda N,Mizuguchi M,Tu lapurkar A A,Shin jo T,Shiraishi M,MizukaMi S,Ando Y,Suzuki Y 2009 Nat.Nanotech.4 158
[19]K resse G,Hafner J 1994 Phys.Rev.B:Condens.Matter Mater.Phys.49 14251
[20]PerdeWJP,Burke K,ErnzerhofM1996 Phys.Rev.Lett.77 3865
[21]Ma Y D,Dai Y,Guo M,Yu L,Huang B B 2013 Phys.Chem.Chem.Phys.15 7098
[22]Zhou J 2015 RSC Adv.5 94679
[23]Lu Y H,Ke C M,Fu MM,Lin W,Zhang C M,Chen T,Li H,Kang J Y,Wu Z M,Wu Y P 2017 RSC Adv.7 4285
PACS:63.20.dk,68.43.Bc,75.20.HrDOI:10.7498/aps.66.166301
*Pro ject supported by the National Natu ral Science Foundation of China(G rant Nos.61674124,11304257,11604275,61227009),the National Basic Research PrograMof China(G rant No.2016YFB 0400801),the Natu ral Science Foundation of Fu jian Province,China(G rant Nos.2014J01026,2016J01037,2015J01028),and the FundaMental Research Funds for the Central Universities,China(G rant Nos.20720160122,20720150033,20720160044).
†Corresponding author.E-Mail:zhangcM000@xmu.edu.cn
‡Corresponding au thor.E-Mail:ypwu@xMu.edu.cn
Mod ifi cation of sp in electron ic p roperties of Fen/GaSe Monolayer adsorp tion system∗
Lu Yi-Hong Ke Cong-Ming Fu Ming-Ming Wu Zhi-Ming Kang Jun-Yong Zhang Chun-Miao†Wu Ya-Ping‡
(Fujian Provincial K ey Laboratory of SeMiconductors and App lications,Collaborative Innovation Center for Optoelectronic SeMiconductors and Effi cient Devices,DepartMent of Physics,X iaMen University,X iaMen 361005,China)
7 March 2017;revised Manuscrip t
3 June 2017)
G roup-IIIA Metal-Monochalcogenides have been extensively studied due to their unique optoelectronic and spin electronic p roperties.To realize the device app lications,Modifying theirMagnetic properties is desirable.AtoMic doping and vacancy defects have been proven to produce itinerant ferromagnetisMand half-metallicity in GaSe monolayer.Relatively,theMagneticModification by adsorbing foreign atoMs is rarely reported.Traditional ferroMagneticMaterial,Fe eleMent,possessing high electronic polarizability and high Curie teMperature,becoMes the best option of adsorbate.In thiswork,Fen(n=1,2)atoMs adsorbed GaSemonolayer systeMs are constructed,and the spin electronic properties are systeMatically studied through the density function theory.Based on the geoMetric con figuration of fully relaxed 3×3 GaSe supercell,three high ly symMetrical sites,i.e.,the holloWsite,the top site of Se atom,and the top site of Ga atoMare inspected to search for the stable absorp tion positions of FenatoMs.CoMputation results of adsorp tion energies indicate that the top site of Ga atoMis preferred for single Fe atom,and the holloWsite near the fi rst Fe adatoMis themost stab le site serving as adsorbing the second Fe atom.Based on themost stab le configuration,the spin electronic properties are studied.For the single Fe adsorbed system,the valence band MaximuMMoves toΓpoint,resu lting in a direct-band-gap.The strong orbit coup ling eff ect between Fe adatoMand its nearest Ga and Se atoMs causes un-coincident majority and Minority spin channels.Two iMpurity bands are located near the FerMi level and contribute on ly to theMinority spin channel,producing a half-Metallicity With a 100%spin polarization in the system.Bader charge analysis and spin-resolved partial density of states suggest that the spin polarization isMainly attributed to the transfer of Fe-3d electrons,and the hybridizations of Fe-3d,Se-4p,and Ga-4p states.Charge transfer froMthe Fe adatoMto GaSe generates an n-type doping and an antiferroMagnetic coup ling between Fe and vicinal Ga and Se atoMs.For the two-Fe-atoMs adsorbed GaSeMonolayer,the spin electronic states are found to beMain ly located between the two Fe adatoMs,leading to the reduction of the charge transfer froMFe to GaSe ML.As the original single spin channel turns into two spin channels(Majority spin channel and Minority spin channel)near the FerMi level,the ferroMagnetic coup ling between Fe atoMand the vicinal Se atoMs turn into antiferroMagnetic coup ling and the spin polarization falls to 0%.Therefore,the spin p roperties of GaSemonolayer can be controlled by modifying the number of adsorbed Fe atoMs.These results reveal the forMation and transforMof the spin electronic properties of typical ferroMagnetic/GaSe adsorption system,which off ers some advice for designing and constructing the two-dimensional spin nanostructures.
surface adsorption,density functional theory,half-metallicity,spin property modification
10.7498/aps.66.166301
∗国家自然科学基金(批准号: 61674124,11304257,11604275,61227009)、 国家重点基础研究发展计划(批准号:2016YFB 0400801)、福建省自然科学基金(批准号:2014J01026,2016J01037,2015J01028)和厦门大学校长基金(批准号:20720160122,20720150033,20720160044)资助的课题.
†通信作者.E-Mail:zhangcM000@xMu.edu.cn
‡通信作者.E-Mail:ypwu@xMu.edu.cn
©2017中国物理学会C h inese P hysica l Society
http://Wu lixb.iphy.ac.cn
