氮肥水平对稻田细菌群落及N2O排放的影响*
2017-09-03宋亚娜陈子强
宋亚娜,林 艳, 陈子强
氮肥水平对稻田细菌群落及N2O排放的影响*
宋亚娜,林 艳, 陈子强
(福建省农业科学院生物技术研究所 福州 350003)
作为土壤氮素转化的驱动者, 微生物群落结构关系着稻田氮素利用及温室气体N2O排放等问题。本研究分别基于高通量测序和荧光定量PCR技术, 分析了不同氮肥水平[CK(不施氮)、N(施N 180 kg∙hm-2)、2/3N(施N 120 kg∙hm-2)、1/3N(施N 60 kg∙hm-2)]下稻田细菌群落及硝化反硝化关键微生物功能基因丰度的变化。结果显示: 氮肥水平提高增加了稻田细菌物种丰富度Chao1指数和群落多样性Shannon指数, 改变了细菌群落组成, 其中与硝化作用相关的硝化螺菌门Nitrospirae和嗜酸的醋杆菌门Acidobacteria的相对丰度随氮肥水平提高而增加, 但甲烷氧化菌Methylosinus的相对丰度随氮肥水平提高而降低。氮肥水平对稻田硝化作用关键微生物氨氧化细菌基因丰度的影响较大, 0~5 cm和10~20 cm深度土层中的基因丰度均随氮肥用量增加而提高; 反硝化作用关键微生物功能基因、和的丰度在不施肥处理(CK)中显著低于施肥处理(1/3N、2/3N和N) (<0.05), 但1/3N、2/3N和N处理的稻田基因丰度没有明显差异; 0~5 cm土层中和基因丰度存在随氮肥水平提高而增加的趋势, 10~20 cm土层中基因丰度在2/3N和N处理下显著高于1/3N处理(<0.05)。N处理的稻田N2O排放通量显著高于2/3N及1/3N处理(<0.05), 后者又显著高于CK处理(<0.05)。相关分析结果表明稻田N2O排放通量与0~5 cm土层中硝化螺菌门Nitrospirae相对丰度及10~20 cm土层中基因丰度存在显著相关性(<0.05,=10)。综上所述, 氮肥水平提高增加了稻田细菌群落多样性, 促进了稻田N2O排放, 且本研究稻田中硝化作用微生物群落及丰度变化与稻田N2O排放的关系更为密切。
氮肥水平; 稻田土壤; 微生物群落; 氨氧化细菌基因; 反硝化细菌基因; N2O排放; 高通量测序; 荧光定量PCR
氮肥不仅是提高作物产量的关键因素, 也是决定农田N2O排放量的重要因素。N2O是重要的农业温室气体之一, 旱地和非饱和水稻土是N2O的主要农业排放源, 由于氮肥投入量的增加, 农业N2O的排放量到2030年预计将增加35%~60%[1]。
由微生物驱动的硝化作用和反硝化作用是土壤释放N2O的两个最重要的途径[2-4]。在硝化作用的氨氧化过程中经过一系列反应经由羟胺形成中间产物N2O, 含有氨单加氧酶基因的微生物在这一过程中起主要驱动作用[5]。土壤中的反硝化细菌在将NO3--N或NO2--N还原为N2的过程中产生N2O, 亚硝酸还原酶基因、氧化氮还原酶基因和氧化亚氮还原酶基因是常作为检测环境中反硝化细菌的标记基因[6]。研究表明施用氮肥有助于提高稻田土壤氨氧化细菌基因丰度[7], 但对反硝化细菌各功能基因多样性的影响各不相同。如, 长期施用氮肥明显提高红壤稻田基因群落多样性[8]; 施氮可改变紫潮泥水稻土反硝化基因的丰度, 但并未明显影响红黄泥水稻土反硝化基因丰度[9]。硝化反硝化作用直接关系到稻田N2O的排放, 硝化反硝化微生物各功能基因多样性及丰度的变化与稻田N2O排放的相关性值得深入研究。
在土壤微生物群落研究中, 与一般的DNA指纹图谱技术相比高通量测序技术具有数据产出通量高、信息量大的特点。高通量测序技术的发展实现了大规模土壤微生物基因直接测序, 为土壤微生物物种、结构、功能和遗传多样性的研究提供了丰富的信息[10-11]。高通量测序在稻田细菌多样性的研究中已大量应用, 如, 在生物黑炭对红壤性稻田根际微生物的研究中, 发现一些对植物生长有益的促生菌有增加的趋势[12]; 采用Illumina miseq高通测序技术对细菌的16S保守区进行测序, 发现轮作土壤微生物丰富度(Chao1)远高于连做[13]。应用高通量测序技术必将有助于稻田N2O释放相关微生物多样性的深入研究。
化学氮肥是决定农田N2O排放量的重要因素, 通过对我国单季稻、稻-旱轮作中的水稻及双季稻等378个点位的N2O排放量的估算, 表明氮肥用量对排放结果具有极显著的影响[14], 而且氮肥施用后也会在短期内引起N2O排放量的显著增加[15]。但是在施用氮肥增加稻田N2O排放过程中有关土壤微生物的作用还不十分清楚, 尤其是硝化与反硝化微生物群落结构变化所发挥的作用更需深入探讨。本研究利用最新的分子生物学技术分析了稻田土壤微生物群落结构的变化及其对稻田N2O排放的影响。我们以田间试验为对象, 通过高通量测序和荧光定量PCR技术分析不同氮肥水平下稻田土壤细菌的种类、群落结构、多样性及硝化和反硝化细菌功能基因丰度, 评价氮肥水平对稻田微生物群落结构与功能的影响及其与稻田N2O排放的相关性。为深入探讨稻田氮素地球生物化学循环中的温室气体排放及持续农业发展提供理论依据。
1 材料与方法
1.1 田间试验
本研究在位于福建省福州市前洋村(26°11′N, 119°16′E)的福建省农业科学院试验基地水稻田进行。供试土壤为红壤, 基本理化性状为: 有机质含量15.77 g∙kg-1、pH 5.82、全氮1.28 g∙kg-1、全磷0.27 g∙kg-1、全钾18.35 g∙kg-1、碱解氮153.6 mg∙kg-1、有效磷15.67 mg∙kg-1、速效钾77.98 mg∙kg-1。
依据当地常规的氮肥用量设置N[180 kg(N)∙hm-2]、2/3N[120 kg(N)∙hm-2]、1/3N[60 kg(N)∙hm-2]和对照CK(不施氮磷钾肥)4个处理。氮肥种类为尿素, 磷肥和钾肥种类为过磷酸钙和氯化钾, 全部磷钾肥和1/2氮肥用做基肥, 1/2氮肥在水稻分蘖后期追肥。磷肥和钾肥均分别为50 kg(P2O5)∙hm-2和100 kg(K2O)∙hm-2。每个处理重复3次, 共12个田间小区随机排列, 小区面积20 m2, 种植密度为175 000株∙hm-2。供试水稻品种为‘荟丰优3301’(LHuifengyou3301), 按当地常规进行水稻栽培的水分及田间管理。田间试验于5月初播种, 6月初插秧, 10月初收获。
1.2 土壤样品采集
于水稻生长最旺盛的开花灌浆期, 每个小区随机选5个点采集土壤样品, 为了更好区分表层和根层土壤, 用土钻分别采集0~5 cm和10~20 cm深度的土层土壤, 将5点采集的土壤混合为1个样品。12个小区取得0~5 cm和10~20 cm土层土样各12个, 共24个土壤样品。田间采集的土样放入4 ℃冰盒中保存, 于实验室内将混合后的土壤去除根系、杂草、土壤动物和石块等杂质并混匀后于-70 ℃冷冻保存用于土壤微生物分析。
1.3 土壤微生物DNA提取
土壤微生物总DNA提取采用FastDNA SPIN Kit (For Soil) (QBIOgene)的试剂盒方法, 称取0.5 g解冻后的土壤样品, 按试剂盒的试验步骤进行土壤微生物总DNA提取, DNA样品-20 ℃冰箱保存待用。
1.4 土壤微生物的高通量测序
将冻存的稻田土壤DNA样品用干冰包装后, 送至北京诺禾致源生物信息科技有限公司, 进行微生物16S扩增子高通量测序。利用通用引物338F: ACTCCTACGGGAGGCAGCAG和806R: GGACTAC HVGGGTWTCTAAT对细菌16S的V3-V4区进行扩增[16], PCR采用高保真酶Phusion High-Fidelity PCR Master Mix with HF Buffer 25mL、DMSO 3mL、F/R引物各3mL、DNA模板 10mL、加无菌水至50mL的反应体系。PCR扩增条件为: 98 ℃, 30 s; 30´(98 ℃, 15 s; 58 ℃, 15 s; 72 ℃, 15 s); 72 ℃, 1 min。PCR产物采用Illumina公司MiSeq测序系统上机分析。利用QIIME(version 1.8.0)对原始序列进行分析[17], 所有样本的序列随机抽取到30 000, 然后进行各项分析, 在97%置信度下将每个样品所有的16S rRNA基因序列进行分类, 获得门、纲、目、科和属各水平下的分类单元。同时利用QIIME计算Alpha多样性, 包括物种丰富度Chao1、香农指数Shannon和辛普森指数Simpson等; 并进行样品间差异分析, 通过STAMP生成主成分分析PCA图[18]。
1.5 土壤微生物的荧光定量PCR
利用氨氧化细菌基因及反硝化细菌的和基因的特异引物[6,19-21](表1), 在ABI PRISM7500 Real-Time PCR System扩增仪上进行绝对荧光定量PCR分析。反应体系为25mL, 其中2mL稀释5倍的DNA模板加反应液23mL, 反应液包括12.5mL SYBR Premix Ex TaqTM(2´), 0.5mL ROX Reference DyeⅡ(50´), 前、后引物各1mL(5 pmol∙L-1, 英骏生物工程公司, 上海)和8mL超纯水。标准曲线的建立依照He等[22]的方法。分别对土壤DNA用各个基因的特异引物进行PCR扩增, 将PCR扩增产物克隆到pMD-18(TaKaRa, 大连)载体中, 然后对插入正确片段的克隆进行质粒DNA提取, 用Nanodrop ND-1000 UV-Vis测定质粒DNA浓度, 计算质粒DNA拷贝数, 按10倍梯度稀释质粒DNA制作标准曲线。本试验中和基因的标准曲线范围分别为9.09´108~9.09´104copies∙L-1、3.67´109~3.67´105copies∙L-1、5.24´109~ 5.24´105copies∙L-1和7.19´109~7.19´105copies∙L-1。和基因标准曲线的2均达0.99, 扩增效率分别为1.01、0.98、0.99和1.01。
1.6 N2O排放通量监测
于土壤样品采集当天, 采用静态密闭箱法采集稻田N2O的排放样品。N2O浓度采用气相色谱(Agilent Technologies 7890B GC system, 美国)测定, 检测器是电子捕获检测器(ECD), 检测器温度330 ℃, 气相色谱的分离材料是PQ填充柱, 柱温55 ℃。标准气体由大连大特气体有限公司提供。N2O排放通量计算公式为[23-24]:

表1 荧光定量PCR的引物
=´´/´273/(273+) (1)
式中:为N2O排放通量[g∙(m2∙h)-1];为N2O标准状态下的密度(1.964 kg∙m-3);为经过田间水层高度调整后采样箱顶部距水面的实际高度(m);/为采样过程中采样箱内N2O浓度变化率[L∙(m3∙h)-1], 即罩箱后密闭时间与箱内气体浓度进行线性回归方程的斜率;为采样箱内的平均温度(℃)。
1.7 数据分析
采用Microsoft Excel 2007和SPSS 13.0软件进行数据处理分析和制图, 处理之间的平均值差异采用one-way ANOVA单因素方差分析, 用Excel 2007中的CORREL公式计算相关系数, 以相关系数显著性检验表确定相关系数的显著性水平。
2 结果与分析
2.1 不同施氮水平下稻田细菌群落变化
2.1.1 多样性变化
本研究通过对16S rRNA的V3-V4区的高通量测序共测得有效序列1 998 211条, 平均每个样品的测序深度为83 258条有效序列。测序所得的一些多样性指数如表2所示。Chao1指数表示物种丰富程度, 其值越高表明物种的丰富度越高; Shannon指数(Shannon diversity index)反映样品的多样性程度, 值越高表明群落的多样性越高; Simpsom指数(Simpson diversity index)反映物种的优势度, 数值越高说明多样性越单一[13]。结果表明稻田细菌的菌种丰富度指数Chao1在CK处理下最低, 1/3N处理下即显著提高(<0.05), 2/3N和N处理间差异虽不显著但二者均显著(<0.05)高于1/3N处理; 同时, 稻田土壤细菌群落的Shannon指数随氮肥水平的提高也显著增加, N处理显著(<0.05)高于2/3N和1/3N处理, 后者又显著(<0.05)高于CK处理; 与Shannon指数相对应的Simpson指数则呈现出施用氮肥的3个处理显著(<0.05)低于不施肥的CK处理; 且上述指标在0~5 cm和10~20 cm两个深度土层中都具有相同的变化规律。
2.1.2 群落结构分析
本研究测得的稻田土壤细菌主要菌门包括: 变形菌门(Proteobacteria)、浮霉菌门(Planctomycetes)、硝化螺菌门(Nitrospirae)、厚壁菌门(Firmicutes)、绿弯菌门(Chloroflexi)、放线菌门(Actinobacteria)和醋杆菌门(Acidobacteria)。这7类菌门占样品总菌量的79.60%~89.30%, 平均84.78%。其中变形菌门、醋杆菌门和绿弯菌门在各样品中所占比例较高, 依次均占26.94%、22.08%和18.55%。可见变形菌门、醋杆菌门和绿弯菌门是本研究的稻田土壤微生物中的优势种群。
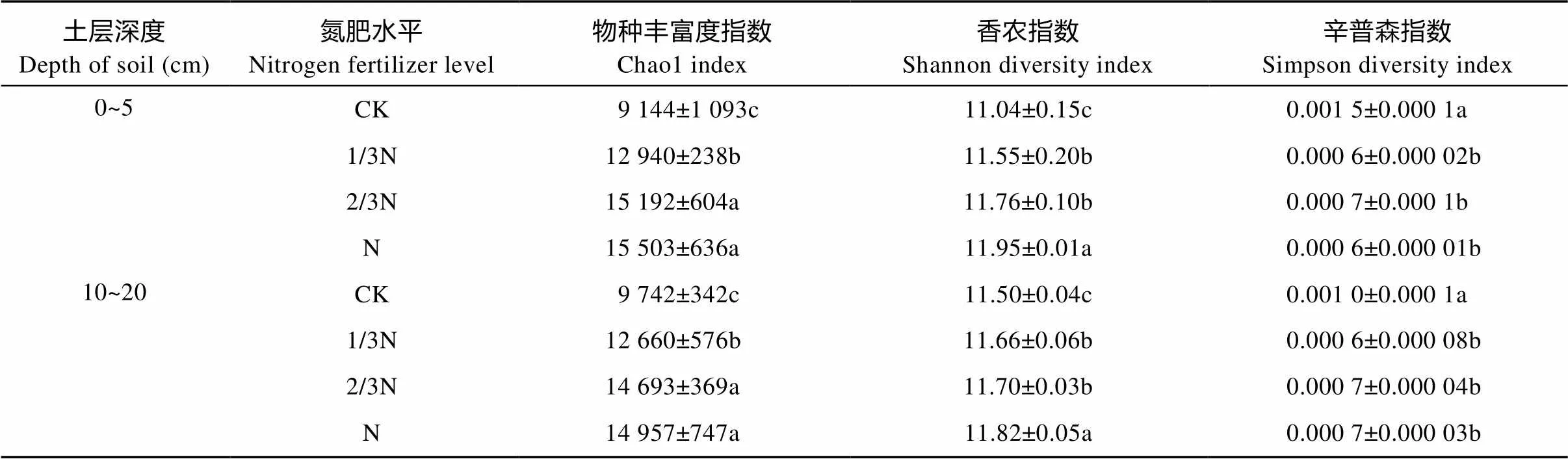
表2 不同氮肥水平下稻田不同深度土壤细菌丰富度及多样性
N: 180 kg(N)∙hm-2; 2/3N: 120 kg(N)∙hm-2; 1/3N: 60 kg(N)∙hm-2; CK: 0 kg(N)∙hm-2. 同一土层同一列数值字母不同差异显著(<0.05)。Values in the same column for the same depth of soil followed by different letters are significantly different (< 0.05).
图1显示了不同处理的土壤细菌各菌门的组成及其平均相对丰度。占土壤细菌总量比例较高的醋杆菌门在0~5 cm和10~20 cm两个土层中均存在随氮肥水平提高其相对丰度逐渐增加的趋势, 相对丰度分别从CK处理的18.40%、17.80%提高到N处理的25.50%、30.00%。不同处理的变形菌门的变化趋势与醋杆菌门相反, 在两个深度土层中其相对丰度分别从CK处理的35.50%、28.20%降低到N处理的22.80%、21.40%。
此外, 不同处理的硝化螺菌门的相对丰度也存在较明显的变化规律, 0~5 cm和10~20 cm两个土层中的硝化螺菌门的相对丰度分别从CK处理的2.80%和3.00%逐渐提高到N处理的5.70%和4.90%。
N: 180 kg(N)∙hm-2; 2/3N: 120 kg(N)∙hm-2; 1/3N: 60 kg(N)∙hm-2; CK: 0 kg(N)∙hm-2.
图2进一步显示了比例最高的前10个属的细菌在0~5 cm(图2a)和10~20 cm(图2b)土层的构成情况。总体而言, 两个深度土层中均存在3个属的菌[互营杆菌()、梭菌属()和甲烷氧化菌()]的相对丰度在氮肥水平提高时降低, 其中甲烷氧化菌的相对丰度在0~5 cm土层中存在明显的N≤2/3N≤1/3N 基于高通量测序的稻田土壤细菌群落组成的主成分分析PCA结果如图3所示。结果显示, CK处理的细菌群落组成与1/3N、2/3N和N处理的细菌群落组成在变异较大的PC1方向上分布差异明显, CK处理单独聚类; 1/3N处理与2/3N及N处理在PC1 方向上也存在一定分布差异; 2/3N处理与N处理在PC1方向上几乎没有分布差异, 仅在变异较小的PC2方向上存在分布差异。此外, CK和1/3N处理的0~5 cm土层细菌群落组成与10~20 cm土层的细菌群落组成在PC2方向上也存在一定差异; 2/3N及N处理的不同深度土层的细菌群落组成几乎没有分布差异。说明随氮肥水平的提高稻田细菌群落组成在逐渐发生变化, 不施氮肥的稻田细菌群落组成与施用氮肥的稻田细菌群落组成差异最大, 在施用氮肥的不同处理间, 中、高氮肥处理的稻田细菌群落组成较为接近, 与低氮处理存在一定差异, 但差异不显著。 2.2 稻田硝化反硝化微生物功能基因丰度分析 利用荧光定量PCR技术分析了与稻田氮素转化及N2O排放相关的关键微生物功能基因的丰度。驱动土壤氮素硝化过程关键步骤的基因丰度如图4a所示。在0~5 cm和10~20 cm两个深度土层中氨氧化细菌基因丰度均随氮肥用量的提高而增加。0~5 cm土层中CK处理的氨氧化细菌基因丰度显著(<0.05)低于1/3N处理, 1/3N处理又显著(<0.05)低于2/3N和N处理; 10~20 cm土层中各个处理间的差异都到达了显著水平(<0.05)。 N: 180 kg(N)∙hm-2; 2/3N: 120 kg(N)∙hm-2; 1/3N: 60 kg(N)∙hm-2; CK: 0 kg(N)∙hm-2. 同一属不同字母表示差异显著(<0.05)。Different letters above bars at the same genus indicated significant difference (< 0.05). N: 180 kg(N)∙hm-2; 2/3N: 120 kg(N)∙hm-2; 1/3N: 60 kg(N)∙hm-2; CK: 0 kg(N)∙hm-2. 同时, 本研究测定了驱动反硝化过程的3个主要微生物功能基因、和的丰度。图4b显示了反硝化细菌基因丰度的变化, 在0~5 cm和10~20 cm两个深度土层中不施肥的CK处理均显著(<0.05)低于其余施肥的处理, 但在1/3N、2/3N和N处理间反硝化细菌基因丰度没有明显差异。结果表明施用氮肥可显著提高稻田土壤硝化细菌基因丰度, 但氮肥用量多少的影响不显著。 不同氮肥水平下的稻田反硝化细菌基因丰度在0~5 cm土层中变化较大, 随氮肥水平提高而增加,呈现N≥2/3N≥1/3N>CK的变化趋势, 其中N处理显著高于1/3N和CK处理(<0.05), 2/3N和1/3N处理又显著高于CK处理(<0.05)(图4c); 而在10~20 cm土层中1/3N、2/3N和N处理间的基因丰度差异不显著, 但三者均显著(<0.05)高于CK处理(图4c); 此外, 各个处理均呈现出0~5 cm土层基因丰度显著(<0.05)高于10~20 cm土层。表明稻田表层土壤中的基因丰度较高且变化较活跃。 稻田反硝化细菌基因丰度的测定结果显示(图4d): 0~5 cm和10~20 cm两个深度土层中均存在N>1/3N>CK的显著变化趋势(<0.05); 对2/3N处理而言, 在0~5 cm土层中处于N和1/3N之间, 与这二者的差异都不显著; 在10~20 cm土层中2/3N处理的基因丰度显著(<0.05)高于1/3N处理, 但与N处理间差异不明显。表明稻田土壤中的基因丰度变化较为活跃。 N: 180 kg(N)∙hm-2; 2/3N: 120 kg(N)∙hm-2; 1/3N: 60 kg(N)∙hm-2; CK: 0 kg(N)∙hm-2. 同一深度土层内各柱上不同字母表示差异显著(<0.05)。Different letters above bars at the same depth of soil indicated significant differences (< 0.05). 2.3 稻田N2O排放状况 本研究在水稻生长最旺盛的开花灌浆期分析了土壤微生物群落的变化, 同时监测了稻田N2O的排放状况。结果显示氮肥水平提高促进了稻田N2O的排放, N处理的N2O排放通量显著(<0.05)高于其他处理, 1/3N和2/3N处理的N2O排放通量差异不显著, 但二者也均显著(<0.05)高于不施肥的对照(CK) (图5)。 将稻田N2O排放通量与土壤硝化反硝化细菌各功能基因拷贝数及硝化螺菌门、属(本研究中比例最高的前10个属中的硝化螺菌4—29和GOUTA19)相对丰度进行相关分析, 结果如表3所示。可见, 0~5 cm土层中硝化螺菌门相对丰度与N2O排放通量的相关系数最高且达到显著性水平(<0.05,=10), 10~20 cm土层中的氨氧化细菌基因拷贝数与N2O排放通量的相关系数最高也达到显著性水平(<0.05,=10)。其余指标与N2O排放通量的相关性不显著。 N: 180 kg(N)∙hm-2; 2/3N: 120 kg(N)∙hm-2; 1/3N: 60 kg(N)∙hm-2; CK: 0 kg(N)∙hm-2. 柱上不同字母表示差异显著(<0.05)。Different letters above bars indicated significant differences (< 0.05). 随着DNA测序技术的发展, 高通量测序技术逐渐成为研究土壤微生物多样性及评价土壤质量的重要手段和方法。本研究利用高通量测序技术分析了不同氮肥水平下稻田土壤细菌群落的变化情况。发现水稻开花灌浆期内, 施肥的稻田细菌物种丰度程度和多样性都明显高于不施肥的稻田, 且氮肥水平提高有助于增加土壤细菌丰富程度, 但当氮肥水平较高后土壤细菌丰富度的增加不再明显。与不施肥的稻田土壤比较, 施肥的稻田土壤养分(速效氮、磷、钾)及有机碳含量都会增加, 可以为细菌生存和生长提供更多的宜居微环境, 使土壤细菌种群多样性增加[25-26]。本试验稻田土壤中变形菌门、醋杆菌门和绿弯菌门是占土壤细菌比例较高的3个主要细菌门。其中醋杆菌门相对丰度受氮肥水平影响最大, 氮肥用量增加促进了醋杆菌门相对丰度的提高。醋杆菌是一类嗜酸的细菌, 氮肥水平提高有助于降低土壤pH增加土壤酸性[7], 从而促进了醋杆菌门相对丰度的增加。 表3 稻田不同深度土层功能基因拷贝数及硝化螺菌相对丰度与N2O排放通量的相关系数 *显著性检验水平达0.05, 自由度为10。*: significant level is up to 0.05, freedom degree is 10. 与氮素转化密切相关的硝化螺菌门相对丰度在稻田细菌中所占比例虽然不高, 但对氮肥水平的提高响应强烈, 其相对丰度随氮肥用量增加而显著提高。同时, 在稻田土壤细菌中所占比例最高的10个细菌属中的4—29和GOUTA19都属于硝化螺菌门, 且氮肥水平提高促进了这二者相对丰度的增加。硝化螺菌是驱动土壤硝化过程的主要微生物, 其相对丰度的提高可能增强稻田硝化作用, 而在硝化作用过程中伴有N2O的释放。相关分析结果显示稻田表层土壤中硝化螺菌门相对丰度与稻田N2O排放量有显著相关性(<0.05,=10)。由此可见, 本研究中硝化螺菌门相对丰度的增加对稻田N2O释放应存在一定贡献。 此外, 在细菌属水平的分析中还发现氮肥水平提高抑制了稻田土壤中的甲烷氧化菌属, 尤其在0~5 cm的表层土中甲烷氧化菌属相对丰度随氮肥水平提高显著降低。这可从微生物群落方面解释氮肥抑制甲烷氧化的机制[27]。 本研究同时利用荧光定量PCR技术分析了稻田土壤中驱动硝化反硝化的微生物功能基因丰度的变化。发现硝化作用限制性步骤氨氧化过程关键酶基因的丰度变化与氮肥用量密切相关, 不同深度土层中均呈现出随氮肥水平提高基因丰度增加的明显趋势, 尤其在深度10~20 cm土中不同氮肥水平处理间的差异都达到显著水平(<0.05)。反硝化作用是在硝酸盐还原酶、亚硝酸盐还原酶、氧化氮还原酶和氧化亚氮还原酶4个酶的连续作用下将土壤中NO3--N或NO2--N还原为N2的过程。对其中3个关键酶基因丰度的测定发现, 不施肥稻田土壤中亚硝酸还原酶基因、氧化氮还原酶基因和氧化亚氮还原酶基因的丰度都显著低于其他施肥处理稻田。而在施肥处理的稻田中,基因和基因的丰度对氮肥用量增加的响应程度明显强于基因, 前2个基因丰度在0~5 cm土层中均存在随氮肥用量提高而增加的趋势, 且10~20 cm土层中的基因在中、高氮处理下显著提高; 但基因丰度对氮肥用量的增加没有明显响应, 高、中、低氮肥水平间没有差异。说明本研究稻田中驱动反硝化过程的氧化氮还原酶基因和氧化亚氮还原酶基因受氮肥用量的影响较大。 硝化和反硝化过程被认为是土壤释放N2O的重要途径, 通过与驱动硝化反硝化的微生物相对丰度及功能基因丰度的相关分析, 发现在本研究中稻田N2O排放量与硝化作用微生物功能基因丰富程度的相关性更高, 表现在0~5 cm土层中的硝化螺菌门相对丰度及10~20 cm土层中氨氧化细菌基因丰度与N2O排放通量的相关性均达到显著性水平(<0.05,=10)。许多试验研究表明反硝化微生物功能基因丰度与土壤N2O释放之间无明显相关性, 如不同季节土豆地反硝化基因(和)丰度变化与N2O释放量无关[28]; 利用实时PCR技术研究发现短期内(72 h)农田土壤反硝化基因()丰度变化和N2O释放量之间没有相关性[29]; 70%WFPS条件下的湿地反硝化基因丰度与N2O释放之间没有明确的相关性[30]。本研究中施用氮肥或氮肥用量增加引起的反硝化微生物、和基因丰度的提高与稻田N2O释放量也都没有相关性。 综上说述, 本研究通过对水稻开花灌浆期内稻田土壤的分析, 发现与无氮和低氮相比较高的氮肥用量能促进稻田细菌群落多样性提高及改变细菌群落组成, 同时可增加稻田温室气体N2O的排放, 且稻田土壤中参与硝化作用的微生物群落组成与丰度的变化与N2O排放增加的关联更为密切。但因为土壤N2O的产生有多种途径, 由多个酶催化进行, 不同途径的不同酶催化反应间存在相互制约或促进的复杂关系。因此, 还需要通过对水稻生长过程中参与土壤N2O产生的关键微生物功能基因多样性及其作用进行系统的分析研究。此外, 从DNA水平研究硝化反硝化微生物基因群落仅能代表微生物的潜在生理功能, 仍需通过RNA水平深入研究硝化反硝化细菌各功能基因多样性与N2O释放的相关性。 [1] FAO. World Agricultural: Towards 2015/2030: An FAO Perspective[M]. Rome: Earthscan Press, 2003: 358–360 [2] Firestone M K, Davidson E A. Microbiological basis of NO and N2O production and consumption in soil[M]//Andreae M O, Schimel D S. Exchange of Trace Gases Between Terrestrial Ecosystems and the Atmosphere. Chichester: Wiley, 1989: 7–21 [3] 朱永官, 王晓辉, 杨小茹, 等. 农田土壤N2O产生的关键微生物过程及减排措施[J]. 环境科学, 2014, 35(2): 792–800 Zhu Y G, Wang X H, Yang X R, et al. Key microbial processes in nitrous oxide emissions of agricultural soil and mitigation strategies[J]. Environmental Science, 2014, 35(2): 792–800 [4] Zhu X, Burger M, Doane T A, et al. Ammonia oxidation pathways and nitrifier denitrification are significant sources of N2O and NO under low oxygen availability[J]. Proceedings of the National Academy of Sciences of the United States America, 2013, 110(16): 6328–6333 [5] Stephen J R, McCaig A E, Smith Z, et al. Molecular diversity of soil and marine 16S rRNA gene sequences related to beta-subgroup ammonia-oxidizing bacteria[J]. Applied and Environmental Microbiology, 1996, 62(11): 4147–4154 [6] Braker G, Tiedje J M. Nitric oxide reductase () genes from pure cultures and environmental samples[J]. Applied and Environmental Microbiology, 2003, 69(6): 3476–3483 [7] Song Y N, Lin Z M. Abundance and community composition of ammonia-oxidizers in paddy soil at different nitrogen fertilizer rates[J]. Journal of Integrative Agriculture, 2014, 13(4): 870–880 [8] 罗希茜, 陈哲, 胡荣桂, 等. 长期施用氮肥对水稻土亚硝酸还原酶基因多样性的影响[J]. 环境科学, 2010, 31(2): 423–430 Lou X Q, Chen Z, Hu R G, et al. Effect of long-term fertilization on the diversity of nitrite reductase genes (and) in paddy soil[J]. Environmental Science, 2010, 31(2): 423–430 [9] 郑燕, 侯海军, 秦红灵, 等. 施氮对水稻土N2O释放及反硝化功能基因(/)丰度的影响[J]. 生态学报, 2012, 32(11): 3386–3393 Zheng Y, Hou H J, Qin H L, et al. Effect of N application on the abundance of denitrifying genes () and N2O emission in paddy soil[J]. Acta Ecologica Sinica, 2012, 32(11): 3386–3393 [10] 楼骏, 柳勇, 李延. 高通量测序技术在土壤微生物多样性研究中的研究进展[J]. 中国农学通报, 2014, 30(15): 256–260 Lou J, Liu Y, Li Y. Review of high-throughput sequencing techniques in studies of soil microbial diversity[J]. Chinese Agricultural Science Bulletin, 2014, 30(15): 256–260 [11] 郑燕, 贾仲君. 新一代高通量测序与稳定性同位素示踪DNA/RNA技术研究稻田红壤甲烷氧化的微生物过程[J]. 微生物学报, 2013, 53(2): 173–184 Zheng Y, Jia Z J. Next generation sequencing and stable isotope probing of active microorganisms responsible for aerobic methane oxidation in red paddy soils[J]. Acta Microbiologica Sinica, 2013, 53(2): 173–184 [12] 陈庆荣, 王成己, 陈曦, 等. 施用烟秆生物黑炭对红壤性稻田根际土壤微生物的影响[J]. 福建农业学报, 2016, 31(2): 184–188 Chen Q R, Wang C J, Chen X, et al. Effect of tobacco stalk-derived biochar on microbes in rhizosphere soil at red paddy fields[J]. Fujian Journal of Agricultural Sciences, 2016, 31(2): 184–188 [13] 张芳, 林绍艳, 徐颖洁. 水稻连作对江苏地区稻田土细菌微生物多样性的影响[J]. 山东农业大学学报: 自然科学版, 2014, 45(2): 161–165 Zhang F, Lin S Y, Xu Y J. The effect of continuous cropping rice on diversity of soil bacteria microbial in Jiangsu Province[J]. Journal of Shandong Agricultural University: Natural Science Edition, 2014, 45(2): 161–165 [14] 廖千家骅, 王书伟, 颜晓元. 中国稻田水稻生长季N2O排放估算[J]. 农业环境科学学报, 2012, 31(1): 212–218Liao Q J H, Wang S W, Yan X Y. Estimation of N2O emissions from paddy fields during rice growing season in China[J]. Journal of Agro-Environment Science, 2012, 31(1): 212–218 [15] Yano M, Toyoda S, Tokida T, et al. Isotopomer analysis of production, consumption and soil-to-atmosphere emission processes of N2O at the beginning of paddy field irrigation[J]. Soil Biology and Biochemistry, 2014, 70: 66–78 [16] Fadrosh D W, Ma B, Gajer P, et al. An improved dual-indexing approach for multiplexed 16S rRNA gene sequencing on the Illumina MiSeq platform[J]. Microbiome, 2014, 2: 6 [17] Caporaso J G, Kuczynski J, Stombaugh J, et al. QIIME allows analysis of high-throughput community sequencing data[J]. Nature Methods, 2010, 7(5): 335–336 [18] Parks D H, Tyson G W, Hugenholtz P, et al. STAMP: Statistical analysis of taxonomic and functional profiles[J]. Bioinformatics, 2014, 30(21): 3123–3124 [19] Braker G, Zhou J Z, Wu L Y, et al. Nitrite reductase genes (and) as functional markers to investigate diversity of denitrifying bacteria in Pacific northwest marine sediment communities[J]. Applied and Environmental Microbiology, 2000, 66(5): 2096–2104 [20] Rotthauwe J H, Witzel K P, Liesack W. The ammonia monooxygenase structural geneas a functional marker: Molecular fine-scale analysis of natural ammonia-oxidizing populations[J]. Applied and Environmental Microbiology, 1997, 63(12): 4704–4712 [21] Throbäck I N, Enwall K, Jarvis Ä, et al. Reassessing PCR primers targeting,andgenes for community surveys of denitrifying bacteria with DGGE[J]. FEMS Microbiology Ecology, 2009, 49(3): 401–417 [22] He J Z, Shen J P, Zhang L M, et al. Quantitative analyses of the abundance and composition of ammonia-oxidizing bacteria and ammonia-oxidizing archaea of a Chinese upland red soil under long-term fertilization practices[J]. Environmental Microbiology, 2007, 9(9): 2364–2374 [23] Zheng X H, Wang M X, Wang Y S, et al. Comparison of manual and automatic methods for measurement of methane emission from rice paddy fields[J]. Advances in Atmospheric Sciences, 1998, 15(4): 569–579 [24] 石生伟, 李玉娥, 李明德, 等. 早稻秸秆原位焚烧对红壤晚稻田CH4和N2O排放及产量的影响[J]. 土壤, 2011, 43(2): 184–189 Shi S W, Li Y E, Li M D, et al. Influence of early rice straw burning on CH4and N2O emissions and grain yield of later rice fields in red soil[J]. Soils, 2011, 43(2): 184–189 [25] 袁红朝, 秦红灵, 刘守龙, 等. 长期施肥对红壤性水稻土细菌群落结构和数量的影响[J]. 中国农业科学, 2011, 44(22): 4610–4617 Yuan H Z, Qin H L, Liu S L, et al. Response of abundance and composition of the bacterial community to long-term fertilization in paddy soils[J]. Scientia Agricultura Sinica, 2011, 44(22): 4610–4617 [26] Hardin G. The competitive exclusion principle[J]. Science, 1960, 131(3409): 1292–1297 [27] 丁维新, 蔡祖聪. 氮肥对土壤氧化甲烷的影响研究[J]. 中国生态农业学报, 2003, 11(2): 50–53 Ding W X, Cai Z C. Effect of nitrogen fertilizers on methane oxidation in soils by methanotrophs[J]. Chinese Journal of Eco-Agriculture, 2003, 11(2): 50–53 [28] Dandie C E, Burton D L, Zebarth B J, et al. Changes in bacterial denitrifier community abundance over time in an agricultural field and their relationship with denitrification activity[J]. Applied and Environmental Microbiology, 2008, 74(19): 5997–6005 [29] Henderson S L, Dandie C E, Patten C L, et al. Changes in denitrifier abundance, denitrification gene mrna levels, nitrous oxide emissions, and denitrification in anoxic soil microcosms amended with glucose and plant residues[J]. Applied and Environmental Microbiology, 2010, 76(7): 2155–2164 [30] Ma W K, Bedard-Haughn A, Siciliano S D, et al. Relationship between nitrifier and denitrifier community composition and abundance in predicting nitrous oxide emissions from ephemeral wetland soils[J]. Soil Biology and Biochemistry, 2008, 40(5): 1114–1123 Effect of nitrogen fertilizer level on bacterial community and N2O emission in paddy soil* SONG Yana, LIN Yan, CHEN Ziqiang (Institute of Biological Technology, Fujian Academy of Agricultural Sciences, Fuzhou 350003, China) Microbial community structures are relevant for the utilization of nitrogen (N) and the emission of nitrous oxide (N2O) in paddy soils. Nitrification and denitrification are the main ways to produce N2O in soils and nitrification bacteria and denitrifying bacteria respectively drive the processes of nitrification and denitrification. In this study, changes in bacterial communities and the abundance of nitrification bacteria or denitrifying bacteria under different nitrogen fertilizer levels [CK (no N fertilization), 1/3N (N application of 60 kg∙hm-2), 2/3N (N application of 120 kg∙hm-2) and N (N application of 180 kg∙hm-2] in paddy soils were analyzed respectively by high-throughput sequencing and real-time PCR. The analysis in abundance of nitrification bacteria or denitrifying bacteria was based on ammonia-oxidizing bacterialgeneand denitrifying bacterialgene orgeneorgene. The results showed that increase in application of nitrogen fertilizer enhanced Chao1 index and Shannon diversity index of bacterial communities and changed the composition of bacterial communities in paddy soil. The relative abundance of Nitrospirae and Acidobacteria increased with improvement of nitrogen fertilizers, while that of Methylosinus decreased with improvement of nitrogen fertilizers in paddy soils. There was greater impact of increased use of nitrogen fertilizer on the abundance ofgene in paddy soils. The abundance ofgene increased with increasing nitrogen application in the 0-5 cm or 10-20 cm depths of soil. The abundances ofgene,gene andgene in no-fertilizer soil (CK) were significantly lower than those in fertilizer soils (1/3N, 2/3N and N) (< 0.05). There was no significant difference in the abundance ofgene among 1/3N, 2/3N and N treatments. However, there was an increasing tendency in the abundance ofgene andgene with increasing application of nitrogen fertilizer in the 0-5 cm depth of soil. The abundance ofgene in the 10-20 cm depth of soil under both 2/3N and N treatments were significantly higher than that in 1/3N treatment (< 0.05). At the same time, the emission of N2O under N treatment was significantly higher than that under 2/3N or 1/3N treatment (< 0.05), and the latter two were also higher than that under CK (< 0.05). Correlation analysis showed that the emission of N2O was markedly correlated with the relative abundance of Nitrospirae in the 0-5 cm depth of soil and the abundance ofgene in the 10-20 cm depth of soil (< 0.05,= 10). In summary, increasing nitrogen application improved the diversity of bacterial communities and the emission of N2O in the studied paddy soils. Also there was a closer correlation between changes in abundance of nitrification bacteria and the emission of N2O. The results suggested that the influence of nitrification bacteria on the emission of N2O in the studied paddy soils was greater than that of denitrifying bacteria. Nitrogen fertilizer level; Paddy soil; Microbial community; Ammonia-oxidizing bacterial gene; Denitrifying bacterial gene; Nitrous oxide emission; High-throughput sequencing; Real-time PCR Feb. 22, 2017; accepted May 24, 2017 S154.3 A 1671-3990(2017)09-1266-10 10.13930/j.cnki.cjea.170146 2017-02-22 2017-05-24 * 福建省公益类科研院所专项(2015R1019-12)、福建省自然科学基金项目(2015J01105)、福建省科技重点项目(2015N0037)和福建省农业科学院PI项目(2016PI-26)资助 * This study was funded by the Special Fund for Research Institutes in Public Interest in Fujian Province of China (2015R1019-12), Fujian Province Natural Science Foundation (2015J01105), Fujian Province Key Projects of Science and Technology (2015N0037) and Fujian Academy Agricultural Science’s PI Projects (2016PI-26). Corresponding author: SONG Yana, E-mail: syana@sina.com 宋亚娜, 研究方向为土壤微生物分子生态。E-mail: syana@sina.com 宋亚娜,林艳, 陈子强. 氮肥水平对稻田细菌群落及N2O排放的影响[J]. 中国生态农业学报, 2017, 25(9): 1266-1275 Song Y N, Lin Y, Chen Z Q. Effect of nitrogen fertilizer level on bacterial community and N2O emission in paddy soil[J]. Chinese Journal of Eco-Agriculture, 2017, 25(9): 1266-12753 讨论和结论

