热可逆自修复聚氨酯弹性体的制备及表征
2017-08-16杨一林王巍巍蒋智杰
杨一林,卢 珣,王巍巍,蒋智杰
(华南理工大学 材料科学与工程学院,广州 510640)
热可逆自修复聚氨酯弹性体的制备及表征
杨一林,卢 珣,王巍巍,蒋智杰
(华南理工大学 材料科学与工程学院,广州 510640)
为探究本征型自修复聚氨酯材料结构与性能的关系,平衡其自修复效率与强度之间的矛盾,采用六亚乙基二异氰酸酯(HDI)三聚体作交联剂,4,4-二氨基二苯二硫醚(AFD)作扩链剂,将可逆双硫键引入聚酯型聚氨酯弹性体中。研究发现:制备的自修复聚氨酯弹性体拉伸强度可达7.7MPa,在60℃,修复时间为24h的条件下,基于拉伸强度的自修复效率高达97.4%;而普通不含有双硫键(只含氢键作用)的弹性体拉伸强度为9.3MPa,在同等条件下的自修复效率为58.0%,表明双硫键的存在使得弹性体自修复效率在原来的基础上提高了67.9%。制备的弹性体具有多次自修复能力,其二次自修复效率为62.3%。
4,4-二氨基二苯二硫醚;六亚乙基二异氰酸酯三聚体;聚氨酯;本征型自修复;拉伸强度;热可逆
自修复高分子材料是一种新兴的智能材料,这类材料能对其自身在成型、加工和使用等过程中产生的内部损伤自发进行部分或完全的修复,从而一定程度上消除材料损伤带来的隐患并延长材料使用寿命[1,2]。聚合物基复合材料自修复方法主要分外援型和本征型两大类,其中本征型自修复是通过材料本体结构设计实现无外加修复剂下的(多次)自修复,随着研究的深入,这种本体固有自修复机制的探究和应用已越来越具有吸引力[3,4]。目前报道的本征型自修复分为可逆(动态)共价键[5-8]和非共价力两种[9-12]作用方式,而这两种价键的引入都势必会削弱材料的力学性能,这也一直是本征型自修复研究与应用之间的矛盾所在。此外,本征型自修复材料理论上应为热固性,同时为了能达到自修复目的材料又应该具有一定流动性,其交联程度不能过高,甚至存在一定缺陷,这一点也加深了材料自修复性能与力学性能之间的矛盾。针对这两点,本工作一方面将芳香族二硫共价键引入聚氨酯体系,其动态二硫交换反应[3,13,14]可以在室温下进行,且无需其他外界条件刺激,为材料温和条件下的自修复提供保障;另一方面从配方设计出发,采用聚酯二元醇作为聚氨酯体系分子软段,HDI三聚体作为交联剂交联部分为硬段,通过配比设计出带有用氨基封端悬挂链的聚氨酯结构,同时促进多重氢键的形成、提高氢键含量。结果表明,实验制得的聚氨酯自修复材料基本实现了室温下自修复,并在同等自修复效率下,其强度是已有研究材料拉伸强度的近9倍[13],初步具有了作为橡胶弹性体使用的潜质。
1 实验
1.1 原料
聚酯二元醇(PBA):工业级(Mn=2088);异佛尔酮二异氰酸酯(IPDI):分析纯;二月桂酸酯二丁基锡(DBTDL):分析纯;六亚乙基二异氰酸酯(HDI)三聚体:分析纯;4,4-二氨基二苯二硫醚(AFD):分析纯;4,4-二氨基二苯二亚乙基:分析纯。
1.2 实验仪器与设备
采用VERTEX70型FT-IR光谱仪,对试样切面采用衰减全反射扫描,扫描范围为4400~450cm-1;采用Viscotek TDA305型凝胶渗透色谱仪,测定聚氨酯预聚物和原料聚酯二元醇的分子量及其分子量分布:以四氢呋喃作为溶剂,在室温下,进样体积100μL,运行时间为50min;采用Z010型电子拉力试验机,对试样进行拉伸力学性能测试:GB/T 528-1998标准、拉伸速率10mm/min。
1.3 聚氨酯预聚物的合成
聚氨酯预聚物的合成(见图1):称取20.1gPBA置于250mL三口烧瓶中,在N2氛围下加入计量好的4.69gIPDI(5%过量)和适量50×10-6的DBTDL,80℃保温4h得到所需的聚氨酯预聚物。

图1 聚氨酯预聚物的反应过程图Fig.1 Schematic illustration of polyurethane prepolymer preparation
1.4 聚氨酯弹性体的合成
在N2保护下,向上述所得聚氨酯预聚体所在三口烧瓶中加入计量好的1.2gHDI三聚体(含量为5%,质量分数,下同),并搅拌,体系开始变得透明澄清状。采用AFD为扩链剂,扩链系数(即扩链剂组分中—NH2摩尔量与预聚物中的—NCO摩尔量之比—NH2/—NCO)取1.4,故称取3.4gAFD溶于3~5mL四氢呋喃中,溶解完全后加入到聚氨酯预聚体与HDI三聚体的混合溶液,保温60℃快速搅拌10min中,在聚四氟乙烯模具中浇注成型。
排气后在60℃保温16h,得到目标聚氨酯弹性体记为弹性体B。制备过程涉及的主线反应方程式见图2。采用4,4-二氨基二苯二亚乙基为扩链剂制得不含双硫键的聚氨酯弹性体作为对照,记为弹性体A。
1.5 聚氨酯弹性体的修复
采用拉伸性能来表征聚氨酯弹性体的自修复性能,每组数据为5根样条测试结果的平均值。(1)将制得的聚氨酯弹性体按规格裁剪成哑铃状(100mm×10mm×1mm)待测试样条,均分为两部分,一部分作为原样,进行拉伸强度测试;(2)另一部分用医用剪刀,从试样颈部中间位置横刀剪断,随后立即拼合,置于模具中;(3)根据实验条件,将试样分别置于不同的时间以及温度下进行自愈合,随后取出并进行拉伸强度测试,记录最大拉伸强度(σ)。将自愈合样条强度与原始样条强度进行比较计算得到自修复效率。自修复效率(H)通常定义[15,16]为:

图2 聚氨酯自修复弹性体的制备过程Fig.2 Preparation process of self-healing polyurethane elastomer
(1)
2 结果与讨论
2.1 聚氨酯预聚物分析
采用FI-IR来跟踪预聚物的合成反应。图3是分别对PBA,IPDI和聚氨酯预聚物进行红外扫描的谱图。3540cm-1是PBA的羟基峰,预聚物中醇羟基峰明显减弱,转而出现很明显的3375cm-1处峰以及新的1527cm-1峰,二者分别为—NH的伸缩振动吸收峰和弯曲振动吸收峰,说明PBA两端的—OH基团与IPDI反应生成了氨基甲酸酯。

图3 PBA,IPDI及聚氨酯预聚物红外光谱图Fig.3 FTIR spectra of PBA,IPDI and polyurethane prepolymer
表1给出了利用GPC测得的聚氨酯预聚物的数均分子量(Mn)及其分布(Mw/Mn)。按照预期, 一分子PBA与两分子IPDI反应,理论上生成的预聚物的Mn应该比PBA的Mn高出444,实际测得高出435,说明反应达到预期,得到了相应的预聚物。

表1 PBA、聚氨酯预聚物的分子量及其分布情况Table 1 Molecular mass and distribution of PBA and polyurethane prepolymer
2.2 聚氨酯弹性体的分析
2.2.1 聚氨酯预聚物反应生成自修复弹性体的红外谱图变化
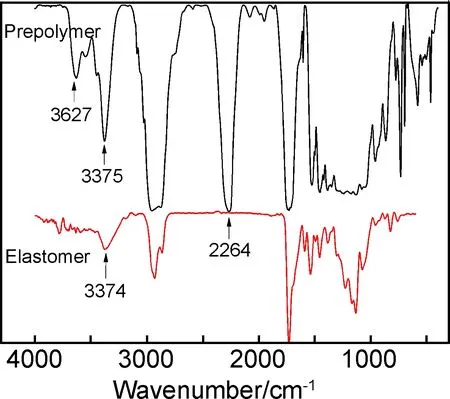
图4 聚氨酯自修复弹性体及其预聚物红外谱图Fig.4 FTIR spectra of polyurethane prepolymer and self-healing polyurethane elastomer
2.2.2 聚氨酯自修复弹性体固化过程中红外谱图的变化
图5为不同反应时间的红外谱图,追踪了聚氨酯弹性体在固化反应生成聚氨酯自修复弹性体的过程。可以看到—NCO特征峰(2266cm-1)的吸收峰随着时间变化峰的强度不断减小,说明—NCO基团随着与—NH2的反应在不断被消耗。从2h到6h内反应速率加快,到6h时—NCO只有少量剩余,24h时已经完全检测不到—NCO基团的存在,表明反应进行完全。扩链剂中—NH2的伸缩振动吸收峰(3462cm-1)随着反应的不断进行不断减弱,但是由于扩链剂过量,可以看到最终有残余的—NH2存在。脲基中的N—H基团伸缩振动峰(3374cm-1)随着反应时间的延长,其吸收强度不断增强,且增强的速度与—NH2消耗的速度基本一致。最后仍有部分—NH2残余,成为氨基封端的支链。
2.3 自修复聚氨酯弹性体结构与性能分析
2.3.1HDI用量对自修复效率的影响
图6研究了HDI三聚体用量对弹性体自修复效率的影响。随着HDI三聚体用量从2.5%增加至12%,聚氨酯自修复弹性体的拉伸强度也相应地从1.9MPa增加至17.5MPa, 而聚氨酯自修复弹性体的自修复效率随着HDI三聚体用量的增加呈现先增加后减小的趋势,在三聚体用量为5%时,弹性体的自修复效率达到最大值97.4%。
为了说明材料交联密度对自修复性能的影响,利用平衡溶胀指数定性表示弹性体交联密度大小。因为硫化橡胶的平衡溶胀指数与其交联密度成反比的关系[18],所以橡胶的溶胀指数越大,其交联密度就越小,相邻的交联点之间的分子链段越长,越有利于分子链段的运动。
HDI三聚体交联剂用量对聚氨酯自修复弹性体的溶胀指数影响如图7所示,弹性体的溶胀指数随着HDI三聚体用量的增加而不断降低,说明对应交联密度不断增高。这与图6中拉伸强度的增加趋势一致,增加的交联密度使得制备的聚氨酯材料硬段含量增加,强度性能提高,但是过高的交联密度会束缚分子链的运动,使得运动链段的自由体积变小,不仅不利于分子链段的运动,还阻碍了分子链中双硫键的动态易位交换作用,因而出现自修复效率的下降。
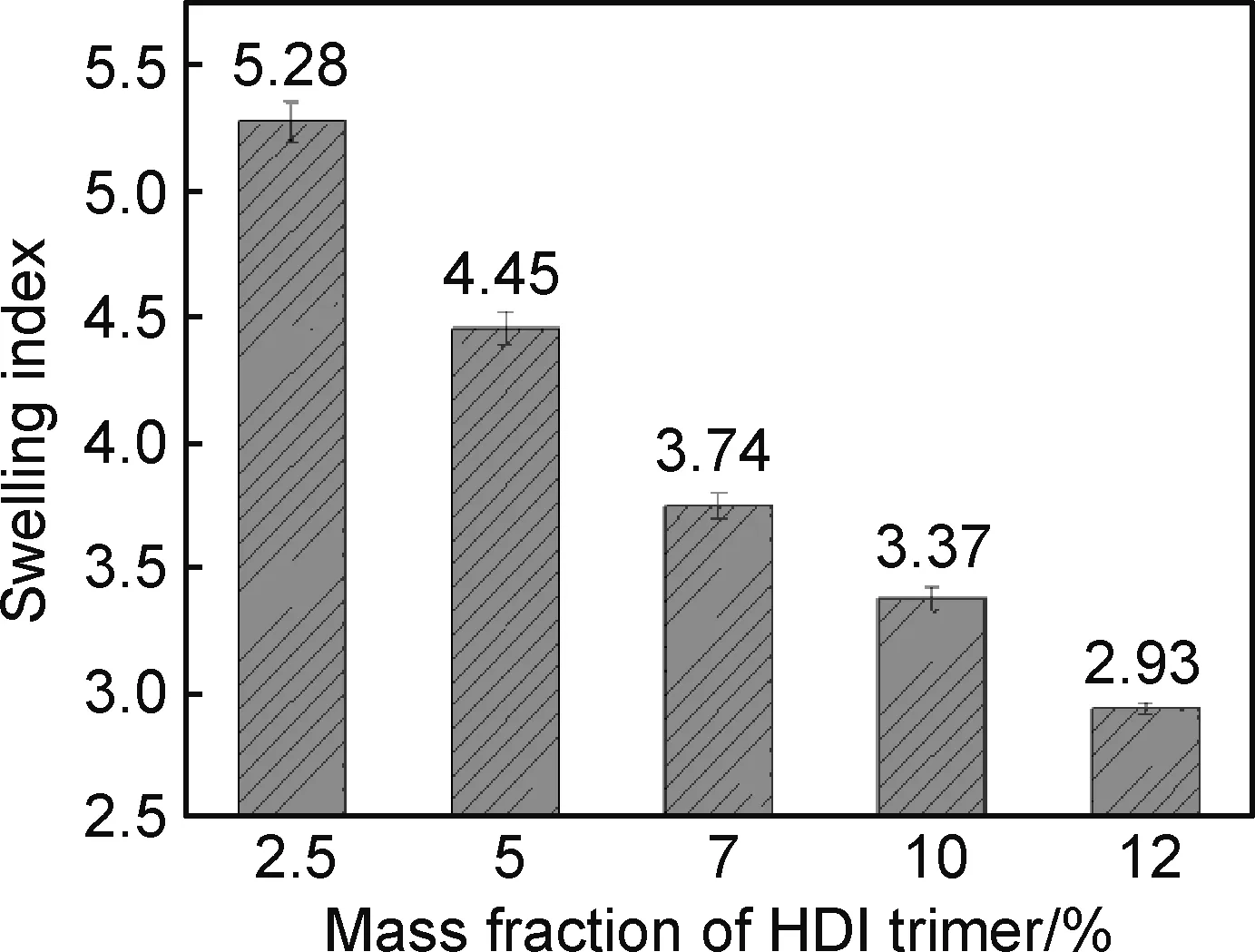
图7 聚氨酯自修复弹性溶胀指数随着三聚体用量的变化Fig.7 Swelling index of PU as a function of HDI trimer content
2.3.2 扩链系数对自修复性能的影响
由于关系到预聚物线性分子之间通过扩链剂产生交联的程度,扩链系数对材料的自修复性能具有重要影响。以R值取2.0、HDI三聚体含量为5%的聚氨酯自修复弹性体作为研究对象,探究了扩链系数(—NH2/—NCO)取值对聚氨酯自修复弹性体的自修复性能的影响。

图8 扩链系数对聚氨酯自修复弹性体自修复性能的影响Fig.8 Self-healing properties of PU as a function of extension coefficient
如图8所示,扩链系数从1.0增加到1.4时,试样的拉伸强度和自修复效率随着扩链系数的增加而增大,这是因为随着AFD扩链剂用量的增加,一方面使聚氨酯硬段含量增加且交联更多,分子间作用力增大,材料的力学性能提高;另一方面扩链剂用量的增加意味着双硫键含量的增加,所以材料的自修复性能逐渐加强,在扩链系数为1.4时材料的自修复效率高达97.4%。当扩链系数从1.4增加至1.8的过程中,弹性体的拉伸强度下降明显,自修复效率也逐渐变低。这主要是因为扩链系数较大时导致反应完成后体系中会有较多AFD剩余。AFD是小分子,混合在体系中会起到增塑剂的作用,所以会显著降低材料的力学性能。
2.3.3 修复温度和时间对自修复效率的影响
通过给予破损弹性体不同修复时间修复,研究了修复时间对材料自修复效率的影响。
由于本实验制备的聚氨酯弹性体是基于双硫键交换反应的,所以其自修复行为可以在室温下进行[13]。图9是弹性体B分别在室温和60℃时的应力-应变曲线,给予材料自修复时间为24h。结果表明材料在室温下基于拉伸强度的自修复效率可达74.0%,表明弹性体B在室温下也具有较好的自修复能力。

图9 聚氨酯自修复弹性体的原样及分别在室温和60℃修复后的应力-应变曲线Fig.9 Stress-strain curves of PU and healed PU treated at room temperature and 60℃
升高温度对于材料中的物理热可逆和化学热可逆都具有促进作用,为提高观察效率在60℃条件下对修复时间进行探究。图10(a),(b)分别是不同修复时间修复后材料的应力-应变曲线和自修复效率图。图10(a)中,随着修复时间从1min增加到12h,修复后材料的应力-应变曲线越来越接近原试样曲线,说明其自修复效率越来越高;对单个曲线而言,随着修复时间从12h增加到24h,修复后材料的应力-应变曲线变化微小,可见材料的自修复在该条件下已经达到一个相对平衡。从图10(b)可以看到,给予材料1min时间的自修复效率不到20%,而有24h修复时间的弹性体自修复效率能高达97.4%,所以材料自修复效率很大程度上受修复时间的影响,在一定时间内,修复时间越长自修复越完整,自修复效率越高,但最终会趋于平衡,达到一个最大自修复效率。
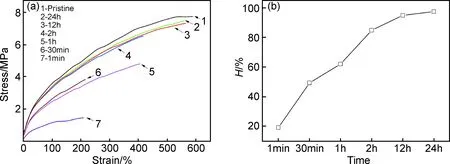
图10 聚氨酯自修复弹性体B在不同时间下的修复(a)应力-应变曲线;(b)自修复效率Fig.10 Polyurethane elastomer B at different healed stages(a)stress-strain curves;(b)healing efficiency
2.3.4 多次自修复效率
双硫键与硫醇基团之间的转化反应可以反复进行,这在理论上支持了双硫键具有多次自修复能力[19,20]。为此,本实验在对修复一次后的样条同一位置再次切断和热处理,得到第二次修复样条,同样处理得到第三次修复样条。随着修复次数增加,试样的拉伸强度从首次修复后的拉伸强度7.5MPa到三次修复后的拉伸强度2.7MPa(见表2),这是因为本身材料在切断受损后,可逆化学键的修复都是具有一定的效率,不能百分百恢复,所以在同一处反复切断,势必会较高程度破坏原有的双硫键及能够形成氢键的结构,因而导致其自修复效率的下降。

表2 自修复效率随着修复次数的变化Table 2 Healing efficiency of samples for different healed times
2.3.5 自修复机理的探究
除了引入的双硫键外,氢键作用[21]对于聚氨酯自修复弹性体也具有重要影响。本部分利用变温红外光谱图跟踪研究了目标弹性体B在不同温度时其基团红外吸收强度的变化,研究了目标弹性体B中氢键的存在与作用机理。图11(a)所示为分别在室温、40℃、60℃、100℃及120℃时对各试样进行红外光谱扫描的结果。一般而言,羰基的特征峰为1670cm-1左右的吸收峰。从图11(b)可看到,在温度从室温变化到100℃乃至120℃的过程中,红外吸收峰的面积减小,同时向高波数移动,游离的羰基被破坏,这与文献[22]中的描述相符。同样,在图11(b)中,随着温度升高,—N—H伸缩振动峰(3374cm-1处)波峰的强度减弱,并且位置在向高波数移动,这说明氢键化的—N—H随着温度的升高不断地往游离的—N—H转变,进一步说明高温破坏了氢键作用。而图11(b)中曲线6是曲线3中弹性体恢复到室温后得到的,可以看到与曲线1相比,各基团的特征峰也基本恢复到未加热时的位置,重新形成了氢键,这表明了体系中的氢键具有热可逆性。

图11 弹性体B的变温红外图谱伸缩振动;(b)N—H伸缩振动Fig.11 FTIR spectra of elastomer B at different temperatures
另外,结合拉伸强度来看,弹性体B结构中存在用氨基封端悬挂链的作用,而悬挂链的存在是使得弹性体B在同等自修复效率下能达到近9倍于已有研究材料[13]拉伸强度的可能原因。图12是以HDI三聚体硬段交联点为代表做出的聚氨酯分子结构局部示意图。
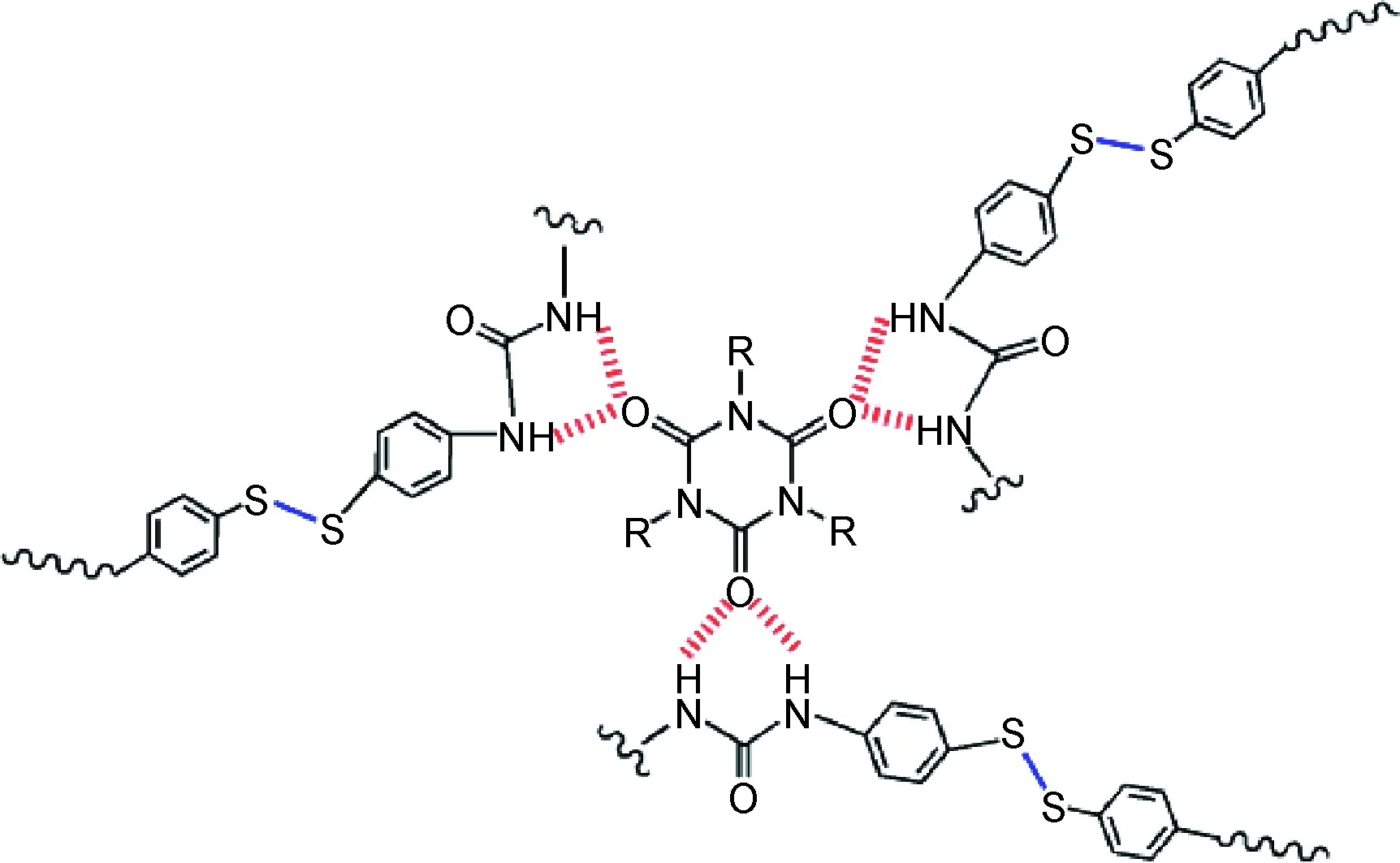
图12 聚氨酯自修复过程中弹性体分子间相互作用Fig.12 Schematic illustration of the hydrogen bonds present in polyurethane elastomer
理论上,扩链系数大于1的时候才会有悬挂链的存在。结合图8可以看到一开始随着扩链系数的增加,拉伸强度也在增加,但当扩链系数超过1.4时,拉伸强度下降,这是因为一味增大扩链系数使得—NH2没有可以反应的位点,造成多余扩链剂小分子残留,降低材料力学性能。扩链系数1.4时悬挂链数量达到饱和,拉伸强度出现最大值。
此外,为了更直观地表示自修复过程,对普通弹性体A和目标弹性体B的哑铃型样条分别从中间部位切断后,人工拼接对齐,在不施加外力的情况下置于60℃下加热24h,并对二者修复前后的拉伸强度进行了测试。从表3中可以看到,弹性体A原样的拉伸强度为9.3MPa高于弹性体B原样的7.7MPa,这是因为后者含有的双硫键键能较碳碳键键能低,所以受到破坏时需要的能量低,故拉伸强度较低;而试样A只存在氢键作用修复后拉伸强度为5.4MPa,反而低于修复后的弹性体B的7.5MPa,二者对应基于拉伸强度的自修复效率分别为58.0%和97.4%。弹性体B的自修复是热可逆的氢键和双硫键共同作用的结果,双硫键的存在使得弹性体自修复效率在只有氢键作用的基础上提高了67.9%。

表3 含双硫键PU弹性体与普通PU弹性体的自修复性能的比较Table 3 Self-healing properties comparison between PU with/without aromatic disulfides
3 结论
(1)采用AFD作为扩链剂、HDI三聚体作交联剂,与PBA和IPDI生成的预聚物反应,成功制备了带有动态易位作用二硫键的自修复聚氨酯弹性体。弹性体的自修复效率随HDI三聚体用量增加、随扩链系数增大都呈现先增大后减小的趋势。
(2)在R值为2.0,HDI用量为5%,扩链系数为1.4的条件下制得的弹性体自修复效率达到最大值。弹性体的室温自修复效率为74.0%;在60℃,修复时间为24h的条件下自修复效率高达97.4%,最大拉伸强度为7.7MPa。所制备聚氨酯弹性体具有多次自修复能力,二次自修复效率为62.3%。
(3)普通不含有双硫键的弹性体在同等条件下的自修复效率为58.0%,双硫键的存在使得弹性体自修复效率在只有氢键作用的基础上提高了67.9%,表明其自修复机理是热可逆的氢键和双硫键共同作用的结果。
[1] 方征平, 羊海棠, 徐立华,等. 聚合物基复合材料自修复体系的构成与修复机制分析[J]. 航空材料学报, 2006, 26(3): 335-336.
FANG Z P, YANG H T, XU L H,et al. Analysis on structure and healing mechanism of self-healing polymer composites[J]. Journal of Aeronautical Materials, 2006, 26(3): 335-336.
[2] BEKAS D G, TSIRKA K, BALTZIS D, et al. Self-healing materials;a review of advances in materials, evaluation, characterization and monitoring techniques[J]. Composites Part B:Engineering, 2016, 87:92-119.
[3] AN S Y, NOH S M, NAM J H, et al. Dual sulfide-disulfide crosslinked networks with rapid and room temperature self-healability[J]. Macromolecular Rapid Communications, 2015, 36(13):1255-1260.
[4] PARK J H, BRAUN P V. Coaxial electrospinning of self-healing coatings[J]. Advanced Materials, 2010, 22(4):496-499.
[5] KUHL N, GEITNER R, BOSE R K, et al. Self-healing polymer networks based on reversible michael addition reactions[J]. Macromolecular Chemistry and Physics, 2016, 217(22): 2541-2550.
[6] JO Y Y, LEE A S, BAEK K Y, et al. Thermally reversible self-healing polysilsesquioxane structure-property relationships based on Diels-Alder chemistry[J]. Polymer, 2017, 108:58-65.
[7] WAN T, CHEN D. Synthesis and properties of self-healing waterborne polyurethanes containing disulfide bonds in the main chain[J]. Journal of Materials Science, 2017, 52(1):197-207.
[8] IDA S, KIMURA R, TANIMOTO S, et al. End-crosslinking of controlled telechelic poly (N-isopropylacrylamide) toward a homogeneous gel network with photo-induced self-healing[J]. Polymer Journal, 2017, 49(2):237-243.
[9] ENKE M, DÖHLER D, BODE S, et al. Intrinsic self-healing polymers based on supramolecular interactions: state of the art and future directions[M]// Self-healing Materials.Switzerland:Springer International Publishing, 2016:59-112.
[10] TEPPER R, BODE S, GEITNER R, et al. Polymeric halogen-bond-based donor systems showing self-healing behavior in thin films[J]. Angewandte Chemie International Edition, 2017, 56(14):4047-4051.
[11] JIANG H, ZHANG G, FENG X, et al. Room-temperature self-healing tough nanocomposite hydrogel crosslinked by zirconium hydroxide nanoparticles[J]. Composites Science and Technology, 2017, 140: 54-62.
[12] MEI J F, JIA X Y, LAI J C, et al. A highly stretchable and autonomous self-healing polymer based on combination of Pt…Pt and π-π interactions[J]. Macromolecular Rapid Communications, 2016, 37(20): 1667-1675.
[13] REKONDO A, MARTIN R, DE LUZURIAGA A R, et al. Catalyst-free room-temperature self-healing elastomers based on aromatic disulfide metathesis[J]. Materials Horizons, 2014, 1(2):237-240.
[14] AMAMOTO Y, OTSUKA H, TAKAHARA A, et al. Self-healing of covalently cross-linked polymers by reshuffling thiuram disulfide moieties in air under visible light[J]. Advanced Materials, 2012, 24(29):3975-3980.
[15] LAI J C, MEI J F, JIA X Y, et al. A stiff and healable polymer based on dynamic-covalent boroxine bonds[J]. Advanced Materials, 2016, 28:8277-8282.
[16] YUAN C, RONG M Z, ZHANG M Q. Self-healing polyurethane elastomer with thermally reversible alkoxyamines as crosslinkages[J]. Polymer, 2014, 55(7):1782-1791.
[17] HOBZA P, REZAC J. Introduction: noncovalent interactions[J]. Chemical Reviews,2016, 116(9):4911-4912.
[18] MEDJNOUN A, BAHAR R. Analytical and statistical study of the parameters of expansive soil[J]. International Journal of Environmental, Chemical, Ecological, Geological and Geophysical Engineering, 2016, 10(2):230-233.
[19] TOBOLSKY A V, MACKNIGHT W J, TAKAHASHI M. Relaxation of disulfide and tetrasulfide polymers[J]. The Journal of Physical Chemistry, 1964, 68(4):787-790.
[20] OWEN G D T, MACKNIGHT W J, TOBOLSKY A V. Urethane elastomers containing disulfide and tetrasulfide linkages[J].The Journal of Physical Chemistry,1964,68(4):784-786.
[21] CORDIER P, TOURNILHAC F, SOULIÉ-ZIAKOVIC C, et al. Self-healing and thermoreversible rubber from supramolecular assembly[J]. Nature, 2008, 451(7181):977-980.
[22] WOLINSKA-GRABCZYK A, KACZMARCZYK B, JANKOWSKI A. Investigations of hydrogen bonding in the poly (urethane-urea)-based membrane materials by using FTIR spectroscopy[J].Polish Journal of Chemical Technology,2008,10(4):53-56.
(本文责编:解 宏)
Preparation and Characterization of Thermally Reversible Self-healing Polyurethane Elastomer
YANG Yi-lin,LU Xun,WANG Wei-wei,JIANG Zhi-jie
(School of Materials Science and Engineering,South China University of Technology,Guangzhou 510640,China)
In order to investigate the structure and property relationships of intrinsic self-healing polyurethane and balance the seemly contradictory forces between its self-healing efficiency and mechanical strength, the reversible disulfide bonds were introduced into polyester-polyurethane by taking hexamethylene diisocyanate (HDI) trimers as the cross-linker and 4,4-diamino diphenyl disulfide as the chain-extender. The results show that the optimal self-healing elastomer exhibits a tensile strength of 7.7MPa and a maximum self-healing efficiency of 97.4% at 60℃after 24 hours, whereas the common elastomer synthesized without disulfide bonds (viaH-bonding interactions) only exhibits a tensile strength of 9.3MPa and a maximum self-healing efficiency of 58.0% under the same condition, indicating that the existence of disulfide bonds helps to increase the self-healing efficiency by 67.9%. The prepared elastomer is found to have multi time self-healing capabilities and the second time self-healing efficiency is 62.3%.
4,4-diamino diphenyl disulfide; hexamethylene diisocyanate trimer; polyurethane; intrinsic self-healing; tensile strength; thermally reversible
10.11868/j.issn.1001-4381.2016.000913
TQ333.99
A
1001-4381(2017)08-0001-08
国家自然科学基金资助项目(51103048)
2016-07-27;
2017-04-20
卢珣(1969-),男,副教授,博士,研究方向:本征型自修复材料研究,通讯地址:广东省广州市天河区五山路381号华南理工大学25号楼442(510640),E-mail:luxun@scut.edu.cn
