三氧化钨表面氢吸附机理的第一性原理研究∗
2017-08-12姜平国汪正兵闫永播
姜平国 汪正兵 闫永播
(江西理工大学冶金与化学工程学院,赣州341000)
三氧化钨表面氢吸附机理的第一性原理研究∗
姜平国†汪正兵 闫永播
(江西理工大学冶金与化学工程学院,赣州341000)
(2016年11月14日收到;2017年1月14日收到修改稿)
采用基于密度泛函理论的第一性原理平面波超软赝势方法,在广义梯度近似下,研究了立方WO3,WO3(001)表面结构及其氢吸附机理.计算结果表明立方晶体WO3理论带隙宽度为0.587 eV.WO3(001)表面有WO终止(001)表面和O终止(001)表面两种结构,表面结构优化后W—O键长和W—O—W键角改变,从而实现表面弛豫;WO终止(001)表面和O终止(001)表面分别呈现n型半导体特征和p型半导体特征.分别计算了H原子吸附在WO终止(001)表面和O终止(001)表面的H—O2c—H,H—O2c···H—O2c,H—O1c—H和H—O1c···H—O1c四种吸附构型,其中H—O1c—H吸附构型的吸附能最小,H—O键最短,H失去电子数最多,分别为−3.684 eV,0.0968 nm和0.55e,此吸附构型最稳定.分析其吸附前后的态密度,带隙从吸附前的0.624 eV增加到1.004 eV,价带宽度基本不变.H的1s轨道电子与O的2p,2s轨道电子相互作用,在−8和−20 eV附近各形成了一个较强的孤立电子峰,两个H原子分别与一个O1c原子形成化学键,最终吸附反应生成了一个H2O分子,同时产生了一个表面氧空位.
第一性原理,三氧化钨,氢气,吸附能
1 引言
三氧化钨(WO3)是一种优良的半导体材料,广泛应用于化学催化、高温超导、气体传感、电致变色、光降解、新能源等领域[1−9],同时也是制取超细钨粉的原料[10,11].钨具有优异的物理和化学特性,如高熔点、高硬度、高密度、高弹性模量、优良的断裂韧性及良好的导热性等性质,是生产多种重要功能和构型材料的主要原料,因而被广泛用于硬质合金、钨基高密度合金、钨丝、钨电极等众多领域[12−14].随着现代工业技术的发展,传统钨粉末制备而成的钨制品已难以满足工业需要,采用超细钨粉制取的钨制品的强度、韧性均有提高,金属塑性-脆性转变温度降低,大大改善了材料性能[15−18].
近年来,许多研究学者对H2还原WO3制取超细钨粉的热力学和还原工艺进行了大量研究[11,19,20],但对其微观扩散吸附动力学方面的研究鲜有报道,因此有必要对此进行理论研究.
密度泛函理论(density functional theory,DFT)是计算物理和化学的重要工具之一,已经成功应用于固体功能材料的结构和性质[21−24],气体及有机化合物在固体表面的吸附性质[25,26],以及表面微观反应机理[27]的研究中.本文采用基于密度泛函理论的第一性原理方法,对立方晶型WO3,WO3(001)表面结构及表面氢吸附机理进行了理论计算,以期为深入认识WO3晶体特性、WO3(001)表面特性及其与H2分子的反应规律提供有益的理论支持.
2 计算模型与方法
2.1 W O 3结构模型
立方晶体WO3的结构模型如图1所示,立方晶体WO3的空间群是PM-3M[28],由W和O构成八面体,W和O分别处于八面体的中心位置和顶点位置,每个W原子含有6个配位,O原子含有2个配位.
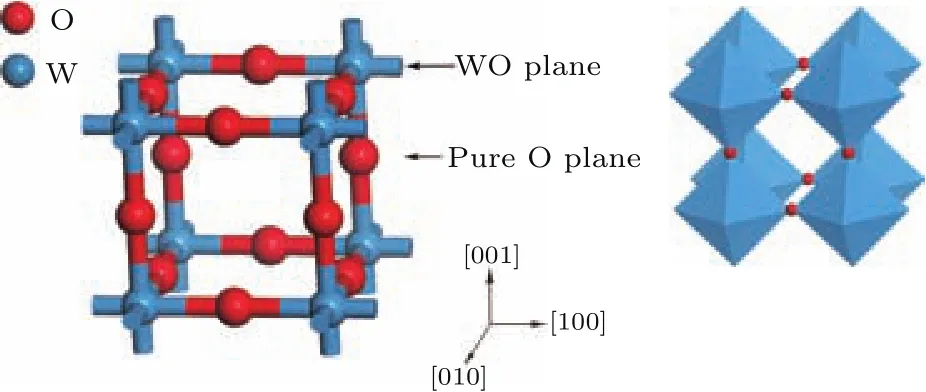
图1 (网刊彩色)WO 3晶体结构示意图Fig.1.(color on line)ScheMatic of WO 3 crystal structure.
2.2 W O 3(001)结构模型
WO3的(001)面表面能最低[29−31],于是创建了两种不同表面的WO3(001)表面结构[32],如图2所示.图2(a)为WO终止(001)表面结构,图2(b)为O终止(001)表面结构;图中W5c和W6c分别为5配位和6配位的W原子,O1c和O2c分别为1配位和2配位的O原子.每个表面结构都含有7层原子,原子层之间设置了厚度为1.0 nm的真空层,真空层厚度和原子层均通过收敛性测试.
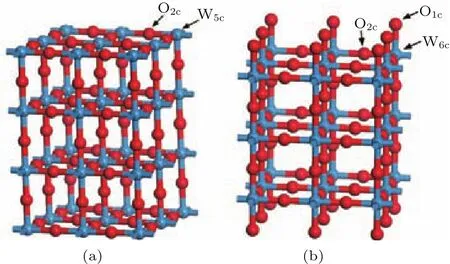
图2 (网刊彩色)WO 3(001)表面结构(a)WO终止(001)表面结构;(b)O终止(001)表面结构Fig.2.(color online)WO 3(001)surface structure:(a)WO-terMinated(001)surface structure;(b)O-terMinated(001)su rface structure.
根据WO3与H2反应的热力学,最终得到W和H2O[11],于是创建图3所示4种不同吸附位置的H原子吸附构型,其中图3(a)和图3(b)分别为WO终止(001)表面结构2个H吸附在同一个O2c位上和两个不同O2c位上,分别记为H—O2c—H和H—O2c···H—O2c;图3(c)和图3(d)分别为O终止(001)表面结构2个H吸附在同一个O1c位上和两个不同O1c位上,分别记为H—O1c—H和H—O1c···H—O1c.O—H原子间距设为0.1110 nm,比水分子在基态时的H—O键长更长.
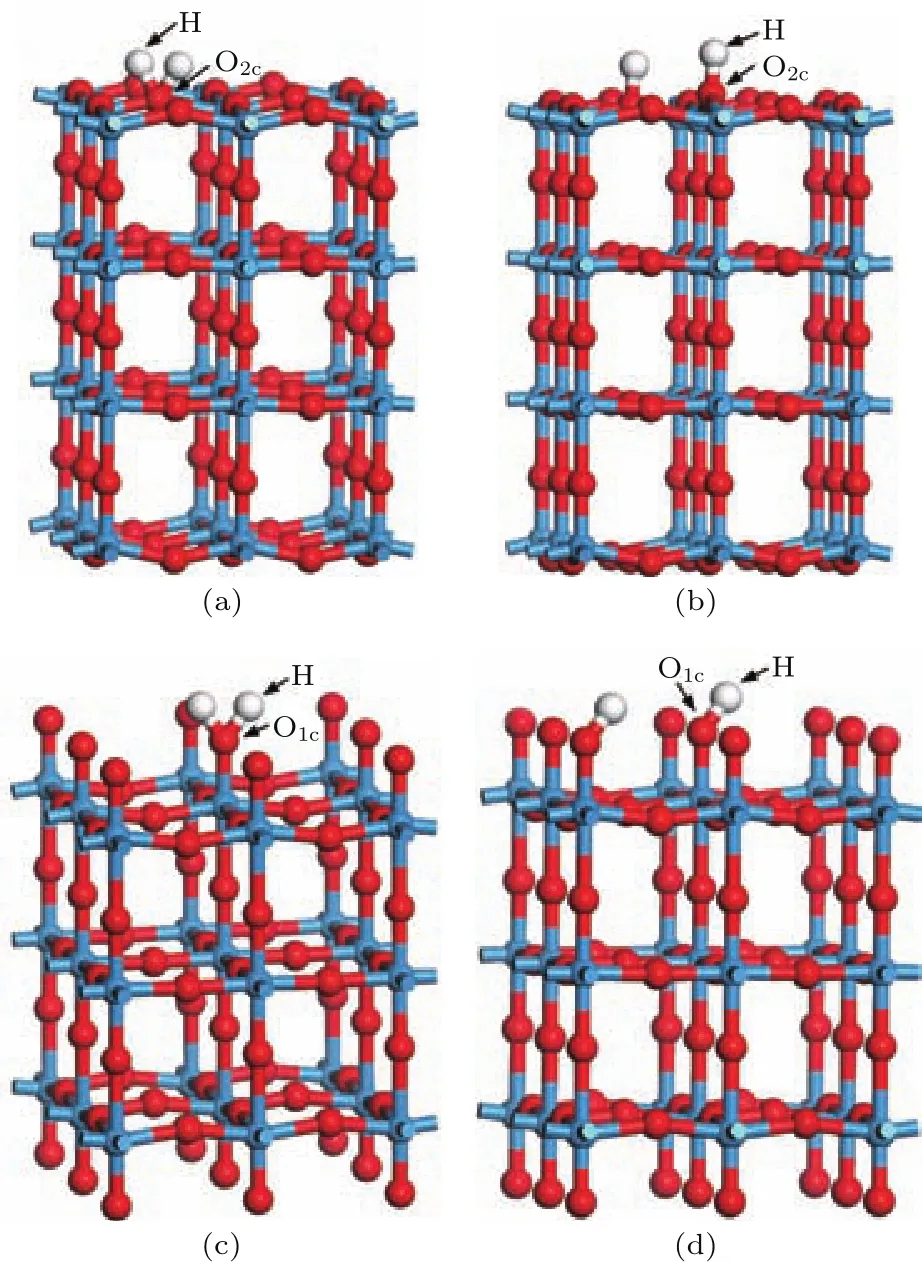
图3 (网刊彩色)WO 3(001)表面吸附H原子构型(a)WO终止(001)表面结构:H—O2c—H;(b)WO终止(001)表面结构:H—O 2c···H—O 2c;(c)O终止(001)表面结构:H—O 1c—H;(d)O终止(001)表面结构:H—O 1c···H—O 1cFig.3.(color on line)H atoMadsorp tion structures on WO 3(001)surface:(a)WO-terMinated(001)su rface structure:H—O 2c—H;(b)WO-terMinated(001)surface structure:H—O2c···H—O2c;(c)O-terMinated(001)su rface structu re:H—O 1c—H;(d)O-terMinated(001)su rface structu re:H—O 1c···H—O 1c.
2.3 计算方法
所有计算工作都由软件Materials Studio中的CASTEP(Cambridge serial total energy package)[33]模块完成,采用广义梯度近似(generalized gradient approximation,GGA)中的PBE(Perdew-Burke-Ernzerhof)方法处理交换关联能[34,35],采用Monkhorst-Pack方法[36]设置K点网格数,运用Vanderbilt超软赝势来处理电子之间的相互作用.对平面波截断能和K点网格数进行收敛性测试,设置平面波截断能为400 eV;将立方晶体WO3的K点网格数设置为8×8×8,将WO3(001)表面结构及其表面吸附H原子结构K点网格数均设置为4×4×1.几何构型优化采用BFGS(Broyden-Fletcher-Goldfarb-Shanno)算法[37],其收敛判别准则和能量计算精度均采用fine,即费米能级(smearing)值为0.1 eV,体系总能量收敛判据为1.0×10−5eV/atom,每个原子上的力收敛判据为0.3 eV/nm,位移收敛判据为0.0001 nm,应力偏差小于0.05 GPa,允许所有原子弛豫.所有计算均在倒易空间中和绝对零度下进行.色散校正和零点能对吸附能的影响较小,忽略二者影响[38].W的原子轨道取为5s25p65d46s2,O的原子轨道为2s22p4,H的原子轨道为1s.
3 计算结果及讨论
3.1 W O 3晶体结构性质
优化立方晶体WO3结构并进行能量计算,其晶格常数a=b=c=0.3838 nm,比实验值0.371 nm[39]略大,且得到图4所示沿着布里渊区高对称点方向的能带结构[40]和图5所示态密度(density of states,DOS).能带结构图和态密度图中将能量零点取为费米面,作为能量的参考点.
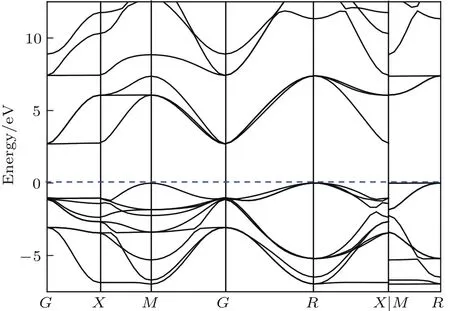
图4 立方WO 3能带结构Fig.4.Band structu re of cubic WO 3.
从图4的能带结构可以看出导带底端是XG,价带顶端是M-R.理论计算带隙宽度Eg为0.587 eV.由于运用密度泛函理论计算会低估Eg,所以Eg值低于实验值2.70 eV,但与张跃[32],Yakovkin等[22]的计算结果基本一致.经剪刀算符对Eg进行修正,可得到WO3带隙宽度Eg为2.70 eV.
由图5的总态密度(total density of states,TDOS)和分态密度(partial density of states,PDOS)可以看出,WO3导带主要来自O-2p和W-5d的电子贡献.WO3价带主要由三部分组成:−7.35—0 eV的上价带主要是O-2p和部分W-5d的电子贡献;−18.5—−16.5 eV范围内的价带主要由O-2s态和少量W-5d态形成;下价带−40—−38 eV范围内还存在一部分孤立能带,这是由W-5p态形成的.
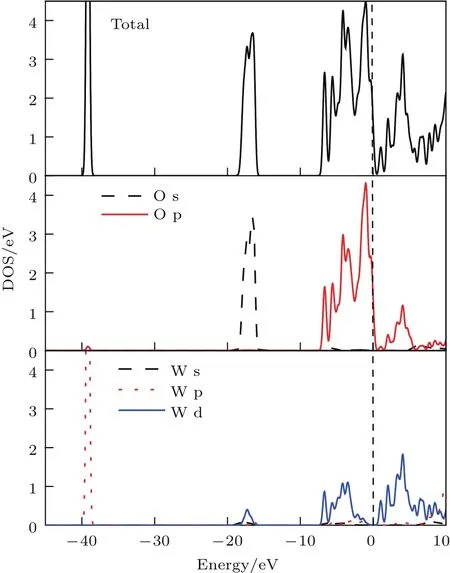
图5 (网刊彩色)立方WO 3总态密度和分态密度Fig.5.(color on line)TDOS and PDOS of cubic WO 3.
3.2 W O 3(001)表面原子结构和电子结构
图6所示为WO终止(001)表面和O终止(001)表面原子结构优化结果.从图中可以看出WO终止(001)表面结构的第一层和第七层的O2c原子向外凸出,O终止(001)表面结构第二层和第六层的O2c原子均向内凹陷,这是由于O原子之间的库仑排斥作用引起的.WO终止(001)表面原子结构中,表面的O2c原子相对于W5c向外凸出,于是形成图6(a)所示锯齿状的W—O—W链型结构,W—O—W键角分别为156.401◦和155.737◦.O终止(001)表面原子结构中,表面的O1c原子向外侧移动,使得与其相连的W6c也向外侧移动,在(001)方向上形成长短相间的W—O键,同样形成图6(b)所示的锯齿状W—O—W链型结构,W—O—W键角分别为165.124◦和165.433◦.库仑力的作用使得WO终止(001)表面和O终止(001)表面锯齿状W—O—W链型结构处的W—O键长都比优化前的键长(0.1919 nm)更长一些.这些现象表明W—O键长和W—O—W键角的改变是表面原子弛豫的主要方式.
图7所示为WO终止(001)表面结构和O终止(001)表面结构优化后的态密度图.从图7可见,WO终止(001)表面结构的导带由W-5d和O-2p电子贡献,费米能级处于导带内,导带内有部分电子填充,呈n型半导体特征[32],附近主要由O-2p电子贡献;价带在−7—−1.75 eV范围内由O-2p和W-5d电子贡献,在−18.5—−15 eV由O-2s和少量的W-5d电子贡献.O终止(001)表面结构的导带主要由W-5d电子贡献,杂化了部分O-2p和微量W-5p电子态,费米能级处于价带内,存在空穴,呈p型半导体特征[32],附近主要由W-5d电子贡献;价带在−9.5—0.3eV范围内由O-2p,部分W-5d和微量W-5p、W-6s电子贡献,在−21—−17.5 eV范围内主要由O-2s电子贡献,杂化了少量W-5d电子与微量W-5p、W-6s电子.
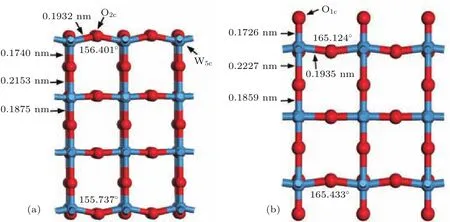
图6 (网刊彩色)WO 3(001)表面弛豫原子结构(a)WO终止(001)表面;(b)O终止(001)表面Fig.6.(color on line)GeoMetrical structure of WO 3(001)surfaces:(a)WO-terMinated(001)surface;(b)O-terMinated(001)su rface.
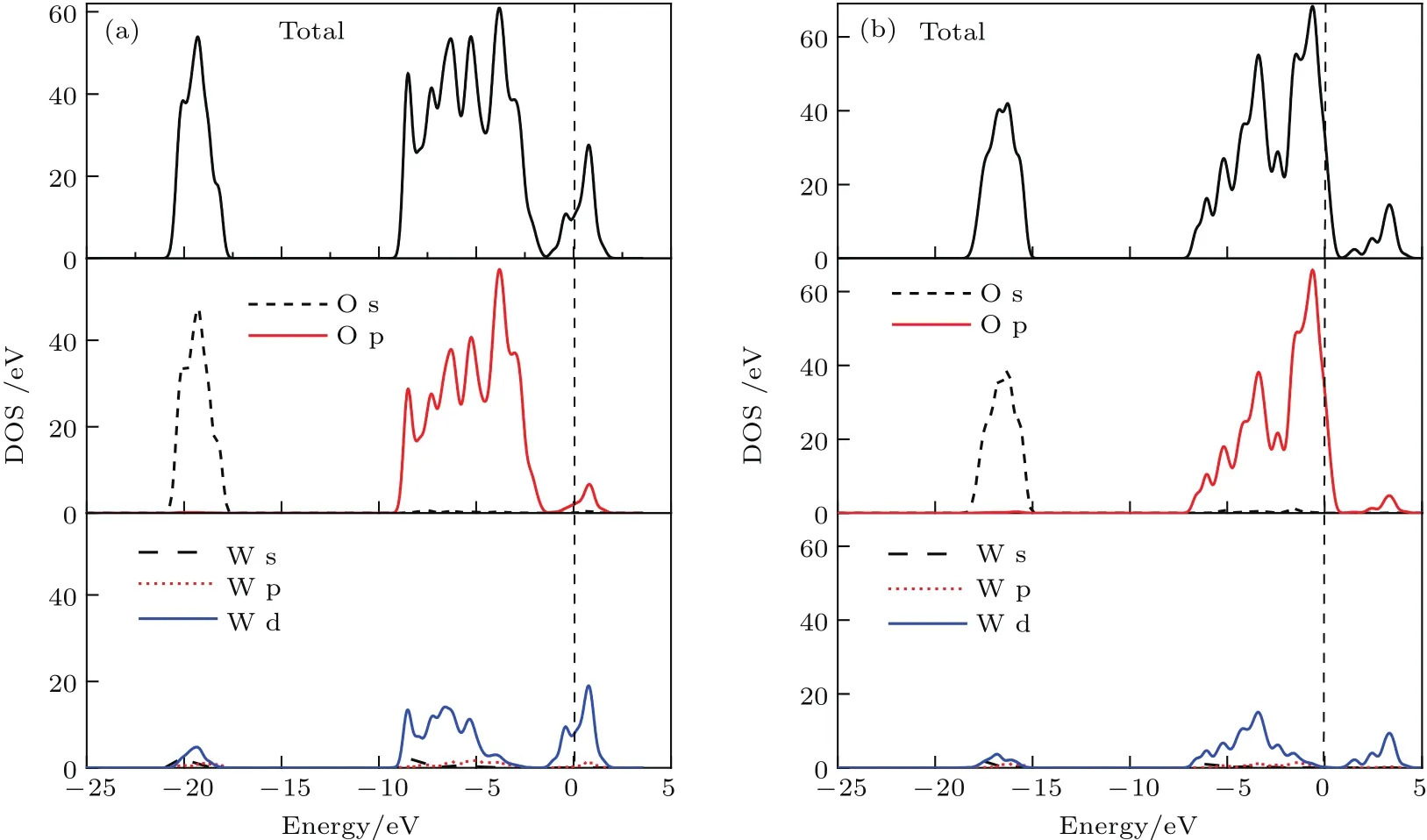
图7 (网刊彩色)WO 3(001)表面结构的总态密度和分态密度(a)WO终止(001)表面结构;(b)O终止(001)表面结构Fig.7.(color on line)TDOS and PDOS ofWO 3(001)surface structu res:(a)WO-terMinated(001)surface structu re;(b)O-terMinated(001)su rface structure.
3.3 H原子吸附在W O3(001)表面
在一定条件下,H原子首先会吸附在WO3表面,然后才会发生化学反应,吸附是发生化学反应的前提.为比较H原子在不同表面不同吸附位置的吸附性能,探讨了图3所示四个吸附位置.
为了探究不同吸附构型的稳定性,首先计算各个吸附构型的吸附能Eads[41]:
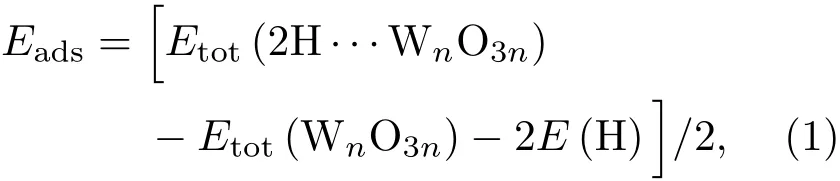
式中Etot(2H···WnO3n)是WO3(001)表面吸附2个H原子后的总能,Etot(WnO3n)是纯净WO3(001)表面结构的总能,E(H)是将一个H原子置于晶格常数为1 nm的立方晶格中计算得到的能量,设置平面波截断能为400 eV,K点网格数为8×8×8,收敛精度为fine,spin设置为2.0,初始自旋值为1µB.
表1所示为四种吸附构型的变化参数,其中吸附能由(1)式计算得到.由表1可以看出四种吸附构型的吸附能均为负值,吸附需要额外的能量,吸附体系比较稳定.对于WO终止(001)表面结构,H—O2c···H—O2c吸附构型最为稳定,吸附能为−3.042 eV,对应的H—O键长为0.0986 nm,Mulliken布居分析表明,H原子带正电荷,为O原子提供了电子.而H—O2c—H吸附构型较为稳定,吸附能为−2.870 eV,形成的H—O键长为0.0994 nm,H原子带正电荷,同样为O原子提供了电子.对于O终止(001)表面结构,H—O1c—H和H—O1c···H—O1c吸附构型都很稳定,吸附能分别为−3.684和−3.133 eV,形成的H—O键长分别为0.0968和0.0979 nm,由于处于O终止(001)表面的最外层O1c原子含一个不饱和化学键,因此易与H原子结合形成H—O键.
图8为WO终止(001)表面结构H原子吸附前后的侧面图.从图中可以看出两个H原子吸附在同一个O2c原子上,使得W—O键长从0.1932 nm增大为0.2168和0.2010 nm,增幅分别为0.0236和0.0078 nm,增幅不大,这是因为W—O键之间作用力较强,W—O键很难打破,致使两个H原子吸附在O原子上难以形成一个H2O分子结构.但Mulliken布居分析表明两个H原子仍为O原子提供了电子,为化学吸附.
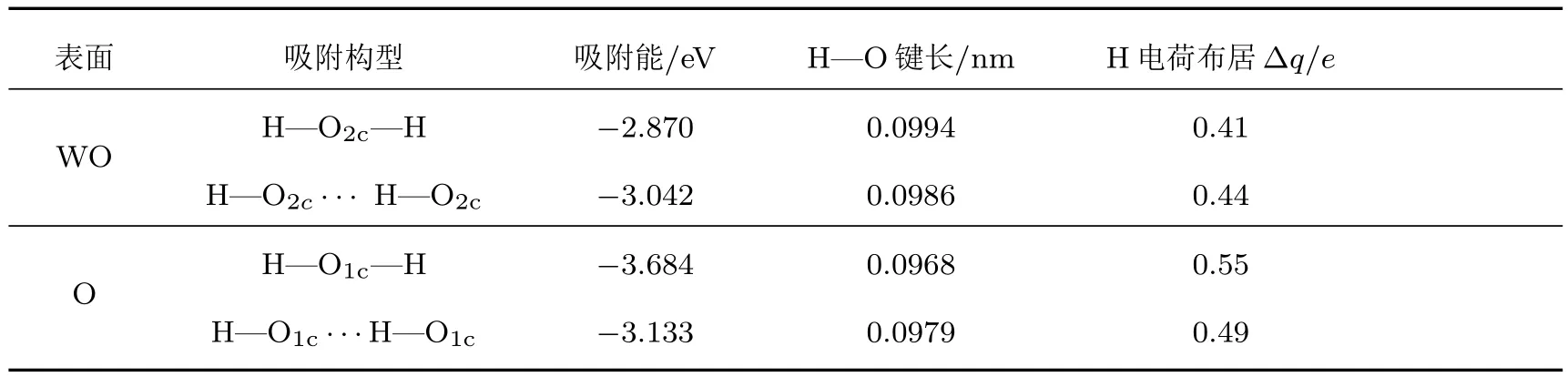
表1 WO3(001)表面吸附H原子后的结构参数变化Tab le 1.Variation of structu ral paraMeters after H atoMadsorp tion on WO 3(001)su rface.
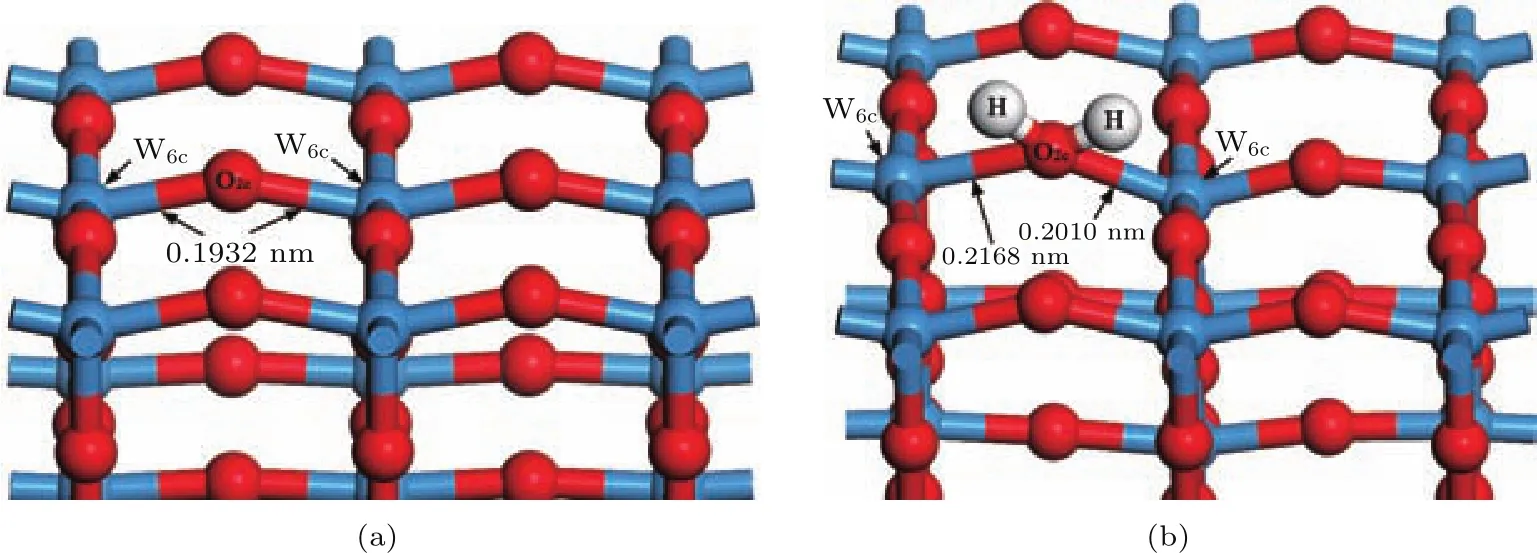
图8 (网刊彩色)WO终止(001)表面H原子吸附前后的结构(a)吸附前;(b)吸附后Fig.8.(color on line)Structures of WO-terMinated(001)su rface(a)before and(b)after H atoMadsorp tion.
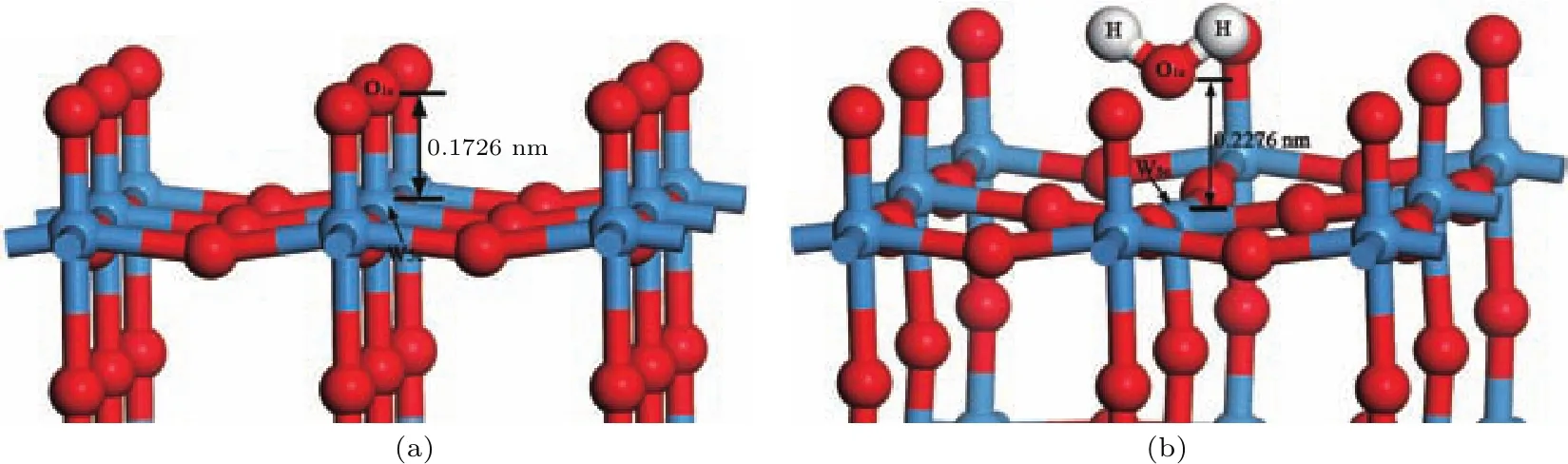
图9 (网刊彩色)O终止(001)表面H原子吸附前后的结构(a)吸附前;(b)吸附后Fig.9.(color on line)Structu res of O-terMinated(001)surface(a)before and(b)after H atoMadsorp tion.
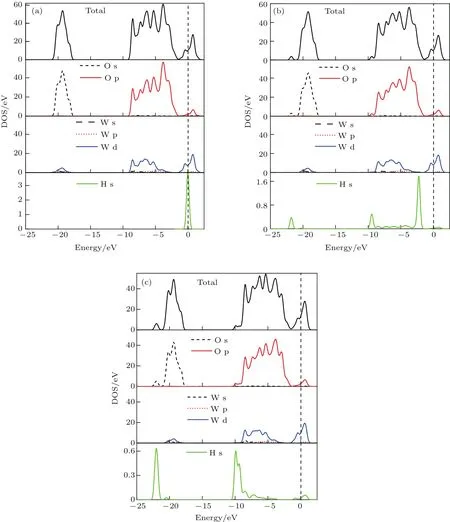
图10 (网刊彩色)WO终止(001)表面H原子吸附前后的总态密度和分态密度(a)WO终止(001)表面H原子吸附前态密度;(b)H—O 2c—H吸附构型的态密度;(c)H—O 2c···H—O 2c吸附构型的态密度Fig.10.(color on line)TDOS and PDOS before and after H atoMadsorp tion on WO-terMinated(001)su rface:(a)DOS before H atoMadsorp tion on WO-terMinated(001)surface;(b)DOS of H—O 2c—H adsorp tion con figuration;(c)DOS of H—O 2c···H—O 2c adsorp tion con figu ration.
图9为O终止(001)表面H原子吸附前后的侧面图.从图中可以看出两个H原子吸附在同一个O1c原子上,使得W6c原子向体相内移动,W—O键长从0.1726 nm增长为0.2276 nm,W—O键断裂,从而在表面顶部形成一个H2O分子,得到的H—O键键长(0.0968 nm)和H—O—H键键角(109.509◦)与水分子在基态时的键长(0.0957—0.1 nm)与键角(104.52◦—109.5◦)基本一致;W—O键的断裂使得W6c原子转变为W5c原子,表面形成了一个氧空位.这种吸附构型基态能量最低,吸附能也最小,是H原子吸附在WO3(001)表面最稳定的吸附构型.
3.3.1 WO终止(001)表面H原子吸附的态密度分布
通过对比H原子在WO终止(001)表面吸附前后的态密度分析了H原子与WO终止(001)表面吸附原子之间的相互作用情况.
图10(a)—(c)分别为吸附前后WO终止(001)表面构型的总态密度、O原子与W原子分波态密度、H原子的态密度.对比图10(a)和图10(b)可以看出H—O2c—H构型吸附前后的态密度发生了变化,H原子吸附到WO终止(001)表面后导致H原子的1s轨道峰向左移动,降低了H原子1s轨道的能量,同时H原子的1s轨道和O原子的2s,2p轨道均产生了较弱的相互作用,sp轨道杂化较弱,在−22.5—−21 eV与−10—−3.5 eV能量区间内形成了较弱的成键电子峰;杂化作用也使得H—O2c—H构型吸附后的总态密度在−22.5—−21 eV与−10—−9 eV能量区间内分别形成了一个较弱的成键电子峰.对比图10(a)与图10(c),同样可以看出H—O2c···H—O2c吸附构型和H原子的态密度都发生了变化,H原子的1s轨道峰同样向左移动,分别与O 2s,2p轨道在−22.5—−21 eV,−10—−8 eV能量区间内发生了轨道重叠,形成了较强的成键电子峰.
3.3.2 O终止(001)表面吸附的态密度分布
图11(a)和图11(b)分别为吸附前后O终止(001)表面H—O1c···H—O1c构型的总态密度、O原子与W原子分波态密度、H原子的态密度.对比两图可以看出吸附后O-2s轨道在−16.3 eV能量处的峰值增大,O-2p轨道在−6.3 eV处峰值增大,在−3.5与−0.75 eV处峰值减小,在−2.5和−5.2 eV处峰值消失.H原子的1s轨道峰向左移动,在−18 eV附近出现了一个孤立峰值,而在−7.5—−2 eV能量区间内连续分布,出现了3个峰值.这表明H与O吸附作用较强,形成H—O化学键,这是由于O终止(001)表面O1c存在不饱和键,易与H结合形成化学键.
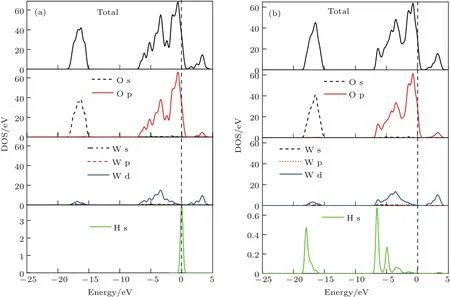
图11 (网刊彩色)H—O 1c···H—O 1c构型H原子吸附前后的总态密度和分态密度(a)吸附前;(b)吸附后Fig.11.(color on line)TDOS and PDOS(a)before and(b)after H atoMadsorp tion on H—O 1c···H—O 1c.
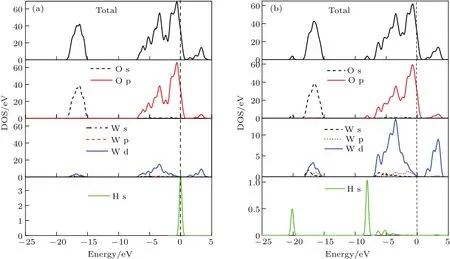
图12 (网刊彩色)H—O 1c—H构型H原子吸附前后的总态密度和分态密度(a)吸附前;(b)吸附后Fig.12.(color on line)TDOS and PDOS(a)before and(b)after H atoMadsorp tion on H—O 1c—H.
H原子吸附在WO3(001)表面不仅影响其表面结构,还会改变其电子结构.图12(a)和图12(b)分别为吸附前后O终止(001)表面H—O1c—H构型的总态密度、O原子与W原子分波态密度、H原子的态密度.从图中可以看出吸附前后总态密度能量值基本不变,导带主要由W-5d电子贡献,杂化部分O-2p电子;价带主要由W-5d,O-2p和O-2s电子贡献.H—O1c—H吸附构型的带隙从吸附前的0.624 eV增加到1.004 eV,价带宽度基本不变.此外,在−8和−20 eV附近处各形成了一个孤立的电子峰,这是由H-1s电子与O-2p,2s电子杂化形成的,致使2个H原子与O1c原子结合形成一个水分子结构.
基于以上分析,H原子吸附在WO3(001)表面最稳定的吸附构型是两个H原子吸附在O终止(001)表面的一个O1c原子上,形成一个H2O分子,与O1c相连的W6c转变为W5c,此表面反应可以用化学方程式表示为[38]
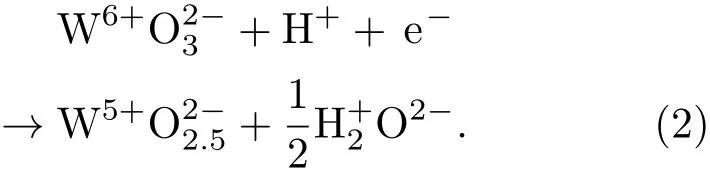
化学方程(2)式很好地反映了WO3氧终止(001)表面的氧空位和电子结构的改变.
根据宏观实验总结出WO3氢还原基本原理[11],用氢还原WO3制取钨粉的总反应为
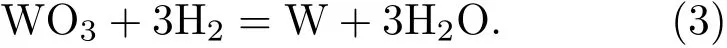
由于钨有四种比较稳定的氧化物,还原反应实际按以下顺序进行:
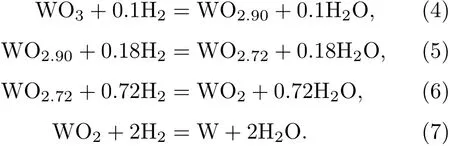
本文通过模拟从微观层面很好地阐述了WO3氢还原反应机理,结合宏观实验更全面地阐述了其反应机理.
4 结论
本文采用基于密度泛函理论的第一性原理方法研究了立方晶体WO3,WO3(001)清洁表面与H原子吸附在WO3(001)表面后的几何结构和电子结构.研究发现WO3(001)表面有WO终止(001)表面和O终止(001)表面两种表面结构,结构优化后表面均形成长短相间的W—O键和锯齿状W—O—W链型结构.H原子吸附在WO3(001)表面最稳定的吸附构型是H—O1c—H,即两个H原子吸附在一个O1c原子上,H的1s轨道与O-2p,2s轨道发生相互作用,H与O形成化学键,生成了一个H2O分子,表面产生了一个氧空位.
[1]Yang Y H,X ie R R,Li H,Liu C J,Liu W H,Zhan F Q 2016 Trans.Nonferrous Met.Soc.China 26 2390
[2]Chen Z,W ang W,Zhu K G 2015 Acta Metall.Sin.28 1
[3]Dai F P,LüS Y,Feng B X,Jiang S R,Chen C 2003 Acta Phys.Sin.52 1003(in Chinese)[代富平,吕淑媛,冯博学,蒋生蕊,陈冲2003物理学报52 1003]
[4]K ukkola J,Mäklin J,Halonen N,K yllönen T,Tóth G,SzabóM,Shchukarev A,Mikkola J P,Jantunen H,Kordás K 2011 Sensor.Actuat.B 153 293
[5]Fang C,W ang H,Shi S Q 2016 Acta Phys.Sin.65 168201(in Chinese)[方成,汪洪,施思齐2016物理学报65 168201]
[6]Zhang T,Zhu Z L,Chen H N,Bai Y,X iao S,Zheng X L,Xue Q Z,Yang S H 2015 Nanoscale 7 2933
[7]Liu X H,Zhou Y,Liang F Y,Qu H N,W en H R 2015 Nonferrous Met.Sci.Eng.6 53(in Chinese)[刘喜慧,周阳,梁福永,曲慧男,温和瑞2015有色金属科学与工程6 53]
[8]Q in Y X,Liu C Y,Liu Y 2015 Chin.Phys.B 24 027304
[9]Zhang F,W ang H Q,W ang S,Wang J Y,Zhong Z C,Jin Y 2014 Chin.Phys.B 23 098105
[10]Vesel A,MozetičM,Balat-Pichelin M2015 Thin So lid FilMs 591 174
[11]Guo F 2007 Mat.Sci.Eng.Powder Metall.12 205(in Chinese)[郭峰2007粉末冶金材料科学与工程12 205]
[12]Li H G,Yang J G,Li K 2010 Tungsten Metallurgy(Changsha:Central South University Press)pp36–39(in Chinese)[李洪桂,羊建高,李昆2010钨冶金学(长沙:中南大学出版社)第36—39页]
[13]Yu G,Han Q G,LiMZ,Jia X P,Ma H A,Li Y F 2012 Acta Phys.Sin.61 040702(in Chinese)[于歌,韩奇钢,李明哲,贾晓鹏,马红安,李月芬2012物理学报61 040702]
[14]Q iu K Q,W ang A M,Zhang H F,Qiao D C,D ing B Z,Hu Z Q 2002 Acta Metall.Sin.38 1091(in Chinese)[邱克强,王爱民,张海峰,乔东春,丁炳哲,胡壮麒2002金属学报38 1091]
[15]Hua J S,Jing F Q,Dong Y B,Tan H,Shen Z Y,Zhou X M,Hu S L 2003 Acta Phys.Sin.52 2005(in Chinese)[华劲松,经福谦,董玉斌,谭华,沈中毅,周显明,胡绍楼2003物理学报52 2005]
[16]Tan J,Zhou Z J,Zhu X P,Guo S Q,Qu D D,Lei MK,Ge C C 2012 Trans.Nonferrous Met.Soc.China 22 1081
[17]Liu H M,Fan J L,Tian JM,You F 2009 China Tungsten Ind.24 29(in Chinese)[刘辉明,范景莲,田家敏,游峰2009中国钨业24 29]
[18]Hessel S,Shp igler B,Botstein O 1993 Rev.Chem.Eng.9 345
[19]W u X W,Luo J S,Lu B Z,X ie C H,Pi Z M,Hu MZ,Xu T,Wu G G,Yu ZM,YiD Q 2009 Trans.Nonferrous Met.Soc.China 19 785
[20]Xu L,Yan Q Z,Xia M,Zhu L X 2013 Int.J.Refract.Met.Hard Mater.36 238
[21]Yu Y X 2013 Phys.Chem.Chem.Phys.15 16819
[22]Yu Y X 2016 J.Phys.Chem.C 120 5288
[23]Yang G M,Xu Q,Li B,Zhang H Z,He X G 2015 Acta Phys.Sin.64 127301(in Chinese)[杨光敏,徐强,李冰,张汉壮,贺小光2015物理学报64 127301]
[24]Xue L,Ren Y M2016 Acta Phys.Sin.65 156301(in Chinese)[薛丽,任一鸣2016物理学报65 156301]
[25]Yu Y X 2014 ACS Appl.Mater.In terfaces 6 16267
[26]Li B,W u T Q,W ang C C,Jiang Y 2016 Acta Phys.Sin.65 216301(in Chinese)[李白,吴太权,汪辰超,江影2016物理学报65 216301]
[27]Gholizadeh R,Yu Y X 2015 Appl.Surf.Sci.357 1187
[28]Chatten R,Chadw ick A V,Rougier A,Lindan P J D 2005 J.Phys.Chem.B 109 3146
[29]Yakovkin IN,Gu tow ski M2007 Surf.Sci.601 1481
[30]Tanner R E,Meethunkij P,A ltMan E I 2000 J.Phys.Chem.B 104 12315
[31]Ma S,Frederick B G 2003 J.Phys.Chem.B 107 11960
[32]T ian X G,Zhang Y,Yang T S 2012 J.Syn.Cryst.41 323(in Chinese)[田相桂,张跃,杨泰生2012人工晶体学报41 323]
[33]Segall MD,Lindan P J D,Probert MJ,Pickard C J,Hasnip P J,C lark S J,Payne MC 2002 J.Phys.Condens.Matter 14 2717
[34]W ang Y,Perdew J P,Chevary J A,Macdonald L D,Vosko S H 1990 Phys.Rev.A 41 40
[35]Perdew J P,Chevary J A,Vosko S H,Jackson K A,Pederson MR,Singh D J,Fiolhais C 1992 Phys.Rev.B 46 6671
[36]Monkhorst H J,Pack J D 1976 Phys.Rev.B 13 5188
[37]F letcher R 1970 CoMput.J.13 317
[38]T ian X G,Zhang Y,Yang T S 2012 Acta Phys.Chim.Sin.28 1063
[39]YaMaguchi O,ToMihisa D,K awabata H,ShiMizu K 1987 J.Am.Ceram.Soc.70 94
[40]Setyawan W,Curtarolo S 2010 CoMput.Mater.Sci.49 299
[41]Sun X,Ku rahashi M,Pratt A,YaMauchi Y 2011 Sur.Sci.605 1067
(Received 14 NoveMber 2016;revised Manuscrip t received 14 January 2017)
PACS:68.43.–h,71.15.Mb,71.20.–b,73.20.AtDOI:10.7498/aps.66.086801
*Project supported by the National Natural Science Foundation of China(Grant No.51564016)and the Natural Science Foundation of Jiangxi Province,China(G rant No.20151BAB206029).
†Corresponding author.E-Mail:pingguo_jiang@163.com
First-p rincip les study on adsorp tion Mechan isMof hyd rogen on tungsten trioxide su rface∗
Jiang Ping-Guo†Wang Zheng-Bing Yan Yong-Bo
(School ofMetallurgy and CheMical Engineering,Jiangxi University of Science and Technology,Ganzhou 341000,China)
W ith the developMent of Modern industrial technology,tungsten products prepared froMnormal tungsten powder cannot meet the demands of industry.The tungsten product produced froMultra-fine tungsten powder exhibits high strength,high toughness,and low Metal p lasticity-brittleness transition teMperature,which greatly iMproves the perforMance of Materials.Hence,it is necessary to carry out theoretical research on the Micro adsorption dynaMics during hyd rogen reduction of tungsten trioxide to p repare u ltra fine tungsten powder.In order to understand crystal characteristics of WO3and WO3(001)surface characteristics,and to provide beneficial theoretical support for reaction law of hydrogen reduction on theWO3(001)surface,theMechanisMs of H atoMadsorption on cubicWO3and WO3(001)surface are studied by the fi rst-p rincip les calculation based on the density functional theory(DFT)p lane wave pseudopotentialMethod.The resu lts show that theoretically calcu lated band gap of the cubic crystalline WO3is 0.587 eV.There are two kinds of WO3(001)surfaces,WO-terMinated(001)surface and O-terMinated(001)surface.The W—O bond length and the bond angle of W—O—W structure change after the geometric optiMization of the surface,and thus the surface relaxation is realized.The WO-terMinated(001)surface show s n-type seMiconductor characteristics while the O-terMinated(001)surface show s p-type seMiconductor characteristics.Four adsorption configurations of H atoMs on the WO-terMinated(001)surface and the O-terMinated(001)surface,including H—O2c—H,H—O2c···H—O2c,H—O1c—H,and H—O1c···H—O1c,are calculated.AMong them,the adsorption energy of the H—O1c—H con figuration is the smallest(−3.684 eV)w ith the shortest bond length of H—O bond(0.0968 nm),and hydrogen atoMs lose theMost of electrons(0.55e),which indicates that the H—O1c—H adsorp tion configuration is theMost stable one.The band gap of the H—O1c—H configuration increases froM0.624 eV to 1.004 eV after adsorption,while the bandw id th of valence band is almost unchanged.The results about the density of states(DOS)reveal that 1s state of the H atoMinteractsw ith 2p and 2s states of the O atom.Strong isolated electron peaks are forMed to be at about−8 and−20 eV.The outerMost O1catoMs of O-terMinated(001)surface contain an unsaturated bond,facilitating the bonding between two H atoMs and one O1catom.Thus,two H atoMs and one O1catoMforMcheMical bonds respectively,and an H2O Molecule is generated,leaving an oxygen vacancy on the surface after adsorption reaction.By combining experiMental observationsw ith simulation results,theMechanisMof hyd rogen reducing tungsten trioxide can be elaborated p rofound ly froMaMicro view.
fi rst princip les,tungsten trioxide,hydrogen,adsorp tion energy
10.7498/aps.66.086801
∗国家自然科学基金(批准号:51564016)和江西省自然科学基金(批准号:20151BAB 206029)资助的课题.
†通信作者.E-Mail:p ingguo_jiang@163.com
©2017中国物理学会C h inese P hysica l Society
http://w u lixb.iphy.ac.cn
