二硫化钼/石墨烯异质结的界面结合作用及其对带边电位影响的理论研究∗
2017-08-12危阳马新国2祝林贺华2黄楚云2
危阳 马新国2)† 祝林 贺华2) 黄楚云2)‡
1)(湖北工业大学理学院,武汉430068)2)(湖北工业大学,太阳能高效利用湖北省协同创新中心,武汉430068)
二硫化钼/石墨烯异质结的界面结合作用及其对带边电位影响的理论研究∗
危阳1)马新国1)2)†祝林1)贺华1)2)黄楚云1)2)‡
1)(湖北工业大学理学院,武汉430068)2)(湖北工业大学,太阳能高效利用湖北省协同创新中心,武汉430068)
(2016年12月15日收到;2017年1月21日收到修改稿)
采用基于色散修正的平面波超软赝势方法研究了二硫化钼/石墨烯异质结的界面结合作用及其对电荷分布和带边电位的影响.研究表明二硫化钼与石墨烯之间可以形成范德瓦耳斯力结合的稳定堆叠结构.通过能带结构计算,发现二硫化钼与石墨烯的耦合导致二硫化钼成为n型半导体,石墨烯转变成小带隙的p型体系.并通过电子密度差分图证实了界面内二硫化钼附近聚集负电荷,石墨烯附近聚集正电荷,界面内形成的内建电场可以抑制光生电子-空穴对的复合.石墨烯的引入可以调制二硫化钼的能带,使其导带底上移至−0.31 eV,提高了光生电子还原能力,有利于光催化还原反应.
异质结光催化,二硫化钼,能带调制,第一性原理
1 引言
随着工业进程的加快,环境污染日益严重,太阳能作为安全无污染的新型能源受到了世界各国的广泛关注.尽管它的应用前景已被广泛接受,但因其传统核心材料较小的光谱响应范围和较低的太阳光利用率使其在实际应用中面临诸多挑战.如传统光催化材料二氧化钛只能吸收波长小于420 nm的紫外光,约占太阳光能量的3%—5%.因此,对于新型光催化材料的寻找迫在眉睫.片状二硫化钼因具有稳定的类石墨层状结构、带隙可调和可见光吸收特性,在催化[1]、太阳能电池[2]、光开关[3]和超级电容器[4]等领域具有广阔的应用前景.尤其是单片层二硫化钼具有较大的比表面积和较多的边缘非饱和键,提供了丰富的活性位点和吸附位点,使其在光催化领域具有极大的应用潜力[5−7].但是单组分二硫化钼光催化剂的能效并不高,主要原因在于较低的载流子传输能力和较高的光生电子-空穴复合率.为了解决这个问题,往往在二硫化钼的纳米体系中引入贵金属、半导体或者共轭聚合物等进行异质结复合,形成肖特基结或者pn结,促进光生电子-空穴的分离,如Ag/MoS2[8],Bi2S3/MoS2[9],Bi2MoO6/MoS2[10],n-rGO/MoS2[11],P3 HT/MoS2[12]等,从而提高二硫化钼的光催化活性.石墨烯具有极大的比表面积、较高的功函数以及优异的导电性能[13],与其他光催化剂复合,能够达到增加光催化剂的吸附位点和抑制电子-空穴复合的双重作用,提升光催化反应效率.Fu和Wang[14]发现铁酸锌/石墨烯复合材料有比纯铁酸锌更高的可见光催化活性,其机理在于石墨烯的引入促进了过氧化氢的生成.Yun等[15]发现钒酸铋和石墨烯复合有效提高了钒酸铋在可见光下催化降解污染物的能力,原因在于钒酸铋受光激发产生的光生电子能持续注入石墨烯中,使得载流子分离,从而提高光生电子的生命周期.此外,Xu等[16]还发现氧化锌/石墨烯复合材料有比纯氧化锌更高的紫外光催化活性,这得益于石墨烯较高的电子迁移率.当前石墨烯与二硫化钼异质结复合的研究已经成为关注的热点之一,主要归因于这种复合可以协同发挥石墨烯高导电性能和二硫化钼光催化活性位点丰富的优势[17−19].实验上往往采用水热法或者原位生长法等[20−22]制备石墨烯/二硫化钼异质结复合体系,探讨界面微结构和表界面光催化反应过程,发现石墨烯作为助催化剂可以提高光催化材料的分散性和产生更多的催化活性点[23],使光量子效率及产氢速率大大提升.
事实上,石墨烯与二硫化钼异质结复合体系的光催化性能及其应用方面取得了重要进展,光量子效率已经超过24%[20],但是大量的研究主要集中在合成工艺、催化活性及性能的表征上,而石墨烯与二硫化钼的结构特殊性使其片层之间可能存在较弱的相互作用,很难通过实验直接进行探测,两者形成的肖特基结[24]或者pn结[21,25]属性也没有形成一致意见,因此有必要对其界面结合作用与电子结构之间的关联性进一步深入研究.采用第一性原理的方法可以获得实验研究无法获取的有用信息,从材料设计的角度分析微结构对光电性能的影响.最近有研究人员采用该方法研究了它们的界面结合及能带结构[24,26],但是有关界面内电荷分布状况以及对能级匹配程度影响的系统研究仍然没有展开,所表现出较强的光吸收能力和较高的载流子迁移率的物理机理仍然不清.为此,我们建立了晶格匹配程度较高的二硫化钼/石墨烯异质结界面模型,采用基于密度泛函理论的第一性原理方法研究了界面结合作用以及其对电荷分布和带边电位的影响,探讨了异质结界面内光生载流子迁移的过程,为进一步改善二硫化钼基光催化剂的光电性能提供理论指导.
2 计算方法和物理模型
采用基于密度泛函理论的平面波超软赝势方法[27]研究了二硫化钼/石墨烯异质结的界面结合作用以及其对电荷分布和带边电位的影响.为了能准确描述层间的范德瓦耳斯力,分别在广义梯度近似(generalized-gradient app roxiMation,GGA)的Perdew-Burke-Ernzerhof(PBE)方案中考虑了Tkatchenko-Scheffl er(TS)和Grimme色散修正[28,29],以及在局域密度近似(local-density approximation)的Ceperley-A lder-Perdew-Zunger方案中考虑了Ortmann-Bechstedt-SchMidt(OBS)色散修正[30].在描述离子实与价电子之间的相互作用时,选取的价电子组态分别为C:2s22p2,S:3s23p4,Mo:4s24p64d55s1.布里渊区k点网格均选取为5×5×4[31].采用Broyden-Fletcher-Goldfarb-Shanno算法对所有模型进行几何结构优化,平面波截断能设置为400 eV,自恰收敛精度设置为5.0×10−5eV/atom,原子间的力场收敛精度设置为1 eV/nm,最大应力设置为0.2GPa,最大位移不超过5×10−4nm.所有计算均由CASTEP软件完成[32].
采用GGA-PBE方法对六方相二硫化钼(空间群:P 63/mMc)的单胞进行几何结构优化,晶格常数为a=b=0.318 nm,c=1.241 nm,同时对石墨烯的单胞也进行几何优化,晶格常数为a=b=0.246 nm,它们与实验值相比误差均小于1%(二硫化钼:a=b=0.316 nm,c=1.230 nm;石墨烯:a=b=0.246 nm)[33,34].具有层状堆叠结构的六方相二硫化钼容易剥离出单层二硫化钼,它是中间一层为Mo原子,上下两层均为S原子的类三明治结构.为了讨论异质结的晶格匹配,建立了两种单层二硫化钼/石墨烯异质结的结构匹配模型.具体为:3×3周期结构的单层二硫化钼超胞与4×4周期结构的石墨烯超胞匹配(图1(a)),以及4×4周期结构的单层二硫化钼超胞与5×5周期结构的石墨烯超胞匹配(图1(b)).图1中片层模型的真空层厚度均选为1.5 nm.
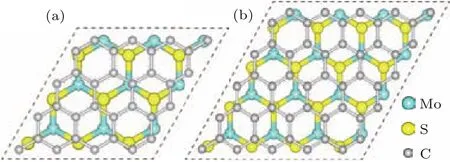
图1 (网刊彩色)两种单层二硫化钼/石墨烯异质结匹配模型的顶视图(a)单层二硫化钼3×3超胞与石墨烯4×4超胞的匹配模型;(b)单层二硫化钼4×4超胞与石墨烯5×5超胞的匹配模型Fig.1.(color on line)Top view s of two Match con figurations ofMonolayer MoS2/graphene heterostructu re:(a)Match con figuration between 3×3 lateral periodicity of Monolayer MoS2 sheet and 4×4 lateral periodicity of graphene;(b)Match con figu ration between 4×4 lateral periodicity ofMonolayer MoS2 sheet and 5×5 lateral periodicity of graphene.
3 结果与讨论
3.1 结构稳定性
为了深入了解异质结的晶格匹配情况,基于图1所示的物理模型,计算了二硫化钼和石墨烯之间的晶格失配率.设定优化后异质结的平衡晶格常数为a′,单层二硫化钼和石墨烯超胞的晶格常数分别为a1和a2,则晶格失配率可以定义为σ=(a2−a1)/a1.由此获得图1中两种异质结匹配形式的晶格失配率分别为3.0%和−3.6%,可见这两种异质结匹配模型均为完全共格(|σ|<5.0%).为了进一步确认异质结的结构稳定性,计算了其晶格失配能.这里晶格失配能定义为

其中E(MoS2)a′和E(graphene)a′分别表示单层二硫化钼和石墨烯的超胞晶格常数为a′时的总能量;E(MoS2)a1表示单层二硫化钼晶格常数为a1时的总能量,E(graphene)a2表示石墨烯晶格常数为a2时的总能量.由此获得两种异质结匹配形式的晶格失配能分别为−0.542和−1.270 eV.事实上,晶格失配能的绝对值越低,异质结的结构越稳定.在随后的研究中,仅选用失配率和失配能均较小的图1(a)异质结模型展开研究.
为了确定二硫化钼和石墨烯之间的层间作用类型,基于图1(a)匹配结构的异质结模型,进一步构建了如图2所示的四种层间对接模型.异质结的界面形成能可以表示为
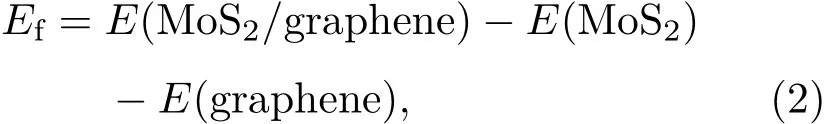
图2 (网刊彩色)二硫化钼/石墨烯异质结的四种层间叠层模型俯视图以及采用三种色散修正方法获得总能量,这里以叠层模型A的总能量为能量参考零点Fig.2.(color on line)Top view sand relative totalenergies using th ree dispersion correction Methods of fou r stacking patterns for MoS2/graphene heterostructu re.The total energy of stacking pattern A is set to zero.
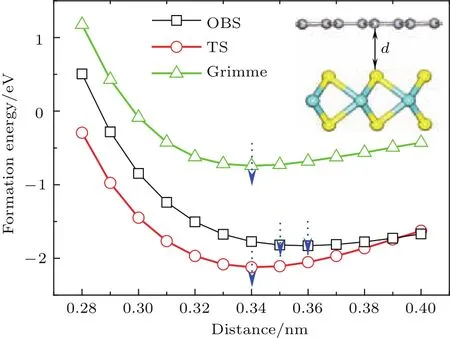
图3 (网刊彩色)采用三种不同色散修正方法计算获得的二硫化钼/石墨烯异质结的层间距d与界面形成能之间的关系Fig.3.(color on line)The relation of interface forMation energy and layer d istance d of MoS2/graphene heterostructu re calcu lated using th ree dispersion correction Methods.
其中E(MoS2/graphene),E(MoS2)和E(graphene)分别表示弛豫二硫化钼/石墨烯异质结、单层二硫化钼和石墨烯的体系总能量.分别采用三种色散修正方法计算了异质结在不同层间距离d下的界面形成能,其关系曲线如图3所示.可以看到TS和Grimme色散修正方法获得最为稳定的层间距d约为0.34 nm,而OBS色散修正方法获得最为稳定的层间距离约为0.35—0.36 nm,这与异质结模型经几何优化后所得最优层间距约0.35 nm结果一致.实验上得到的二硫化钼/石墨烯异质结STEM图谱显示[35],石墨烯与二硫化钼之间最稳定的间距约在(0.34±0.01)nm,这个结果与图3结果相符.三种不同的色散修正方法计算所得异质结形成能随层间距d变化的趋势高度一致,仅能量上稍有差别,这与算法的选取有关.在层间距d约为0.28—0.34 nm时,随着层间距增大,形成能逐步显著降低,由正值变为负值,结构趋于稳定.此时层间相互作用主要表现为斥力,随着层间距增大,斥力减弱,结构趋于稳定;在层间距d约为0.34—0.40 nm时,随着层间距增大,形成能逐步缓慢增加,结构稳定性降低.此时层间相互作用主要表现为引力,随着层间距增大,引力减弱,结构稳定性逐渐降低.这与文献[36,37]结果一致.在四种层间叠层模型中,层间距与形成能的关系曲线趋势均一致,所得最为稳定层间距离的结果也均相同.在此层间距离下,四种叠层模型的界面形成能均为负数,表明层间存在一定的结合作用,使其可以形成较为稳定的异质结.从两个方面可以看出二硫化钼和石墨烯之间存在范德瓦耳斯结合作用.一方面,四种叠层模型的界面形成能的大小非常接近,其差值不超过0.15 eV,表明层间的结合作用与它们层间对接方式并没有太大的关系;另一方面,层间结合作用力均较小,例如图2中模型A的单位面积界面形成能(结合能)仅为1.323 eV/nm2,在典型范德瓦耳斯结合能数值(1.3—2.1 eV/nm2)范围内[38].实验上测量了二硫化钼/石墨烯异质结的光致发光(PL)谱[35],与二硫化钼/二氧化硅的拉曼谱相比,二硫化钼/石墨烯异质结的PL谱有微弱的上移(3 cm−1)和窄化(3 cm−1),由此认为两者之间的相互作用力是范德瓦耳斯作用力.而我们所计算的界面结合能,同样证明了二硫化钼与石墨烯之间存在范德瓦耳斯结合作用.
3.2 能带结构
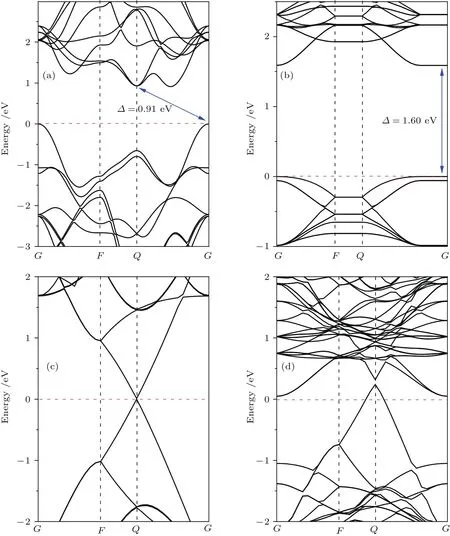
图4 (网刊彩色)体相二硫化钼(a)、单层二硫化钼(b)、石墨烯(c)及二硫化钼/石墨烯异质结(d)的能带结构图Fig.4.(color on line)E lectronic band structures of(a)MoS2 bu lk,(b)MoS2 Monolayer,(c)graphene and(d)MoS2/graphene heterostructure.The FerMi levels are set to zero and Marked by red dotted lines.
为了探究二硫化钼/石墨烯异质结的界面结合作用对界面内电荷分布的影响,这里以图2中模型A为例,计算了异质结的能带结构和态密度.图4为体相二硫化钼、单层二硫化钼、石墨烯以及二硫化钼/石墨烯异质结的能带结构.可以看出,体相二硫化钼显示了高对称G点的价带顶和高对称Q点的导带底之间0.91 eV的间接带隙,而单层二硫化钼显示出高对称G点位置的直接带隙情形,其值为1.60 eV.Low等[39]实验上测得体相和单层二硫化钼的带隙值分别为1.2和1.9 eV;Mak等[40]实验上测得单层二硫化钼的带隙值为1.8 eV;Huang等[41]通过理论计算获得单层二硫化钼的带隙值为1.67 eV.可以发现,我们的计算结果接近其他的理论计算结果,而略低于实验值.由于密度泛函理论在计算带隙值时普遍存在不可避免的误差,GGA近似往往会高估晶格常数并低估带隙值,但不影响能带结构以及其他方面结果的分析.与纯石墨烯的能带结构比较,二硫化钼/石墨烯异质结能带结构中的费米能级发生了移动,使其位于石墨烯狄拉克点的下方,石墨烯导带存在未填充能级,石墨烯形成了带隙值不超过0.1 eV的p型体系.而费米能级位于二硫化钼带隙内的导带底附近,使二硫化钼显示出典型的n型半导体特征.由图4(b)—图4(d)可以看出,石墨烯狄拉克点的位置高于单层二硫化钼导带底的位置,即两者间存在电势差.因此,当二硫化钼与石墨烯形成异质结后,界面电荷将发生相对转移:处于较高位置的石墨烯的电子将流入二硫化钼表面,即二硫化钼表面聚集负电荷,石墨烯表面聚集正电荷,这与计算所得电荷密度差分图(图5)显示的结果一致.由于电荷的重新分布,导致石墨烯电势增加,能带下移;二硫化钼电势降低,能带上移,这也被后面计算出的功函数所证实.
异质结的形成改变了二硫化钼和石墨烯的电学属性,这主要源于层间的结合作用导致的界面内电荷的重新分布.图5(a)显示了二硫化钼/石墨烯异质结的电荷密度差分图,其中红色分布区域表示电子减少(正电荷聚集),蓝色区域表示电子增加(负电荷聚集).可以看出,异质结界面的形成,导致石墨烯上的部分电子向二硫化钼转移,从而使二硫化钼层聚集了较多的负电荷,石墨烯聚集了较多的正电荷,界面内形成了内建电场.二硫化钼/石墨烯异质结界面的能带结构和电荷分布原理如图5(b)所示.Roy等[42]从实验上研究了石墨烯/二硫化钼异质结的光电流效应,当背栅电远小于二硫化钼的传导阈值时,光生电子由二硫化钼向石墨烯转移,光生空穴由石墨烯向二硫化钼转移,这个结果与我们的研究结论一致.
为了更好地了解二硫化钼/石墨烯异质结层间相互作用的微观机理,分析了异质结的总态密度和分态密度,如图6所示.可以看出,在费米能级附近异质结的价带顶主要由Mo 4d和C 2p轨道组成,而导带主要由Mo 4d和S 3p轨道组成.因此,在太阳光照下光生电子可以从价带中Mo 4d轨道跃迁至导带中Mo 4d轨道.从图6(c)中可以看出导带中Mo 4d和S 3p之间存在能级重叠,显示了他们间存在较强的轨道杂化作用,使处于激发状态下的电子很容易从Mo 4d轨道转移至S 3p轨道,激发的电子趋向集中于界面间的S原子层.结合图5可知,在界面内建电场的作用下,S原子附近的光生电子有向石墨烯转移的趋势,从而有利于光生电子-空穴对的有效分离.
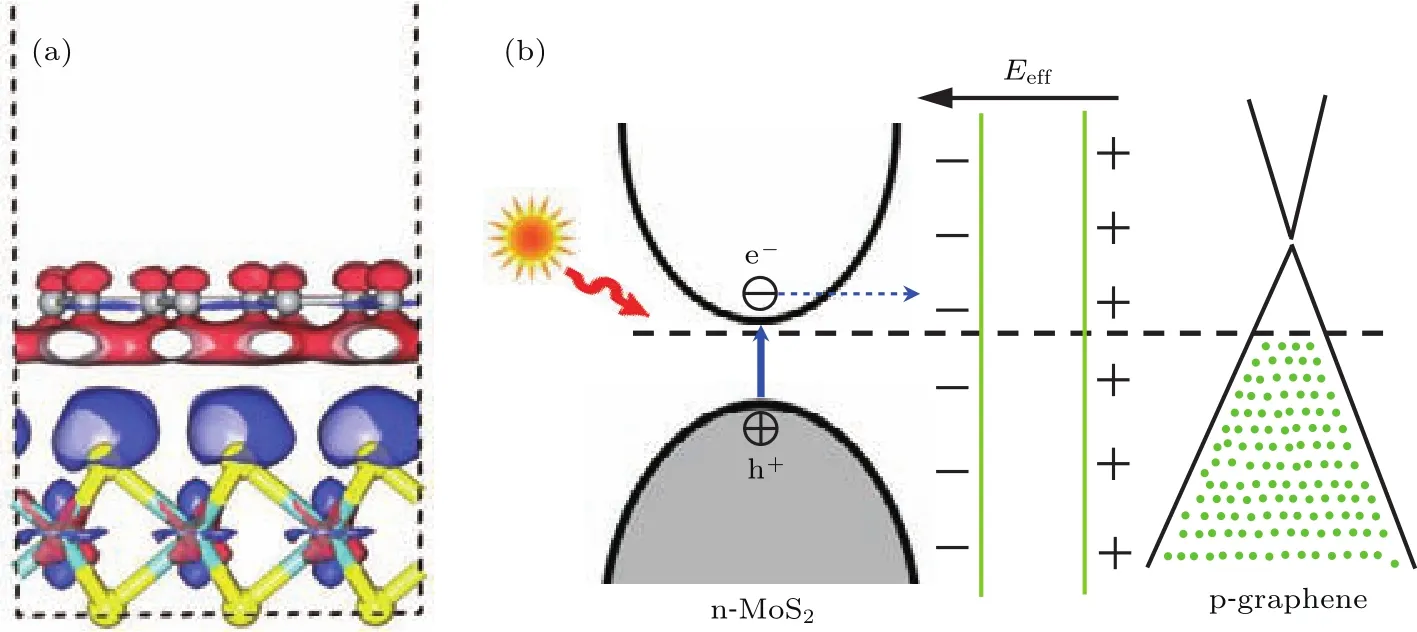
图5 (网刊彩色)二硫化钼/石墨烯异质结的三维电子密度差分图以及界面电荷重新分布原理Fig.5.(color on line)Three-d iMensional charge density d iff erence p lots and p rincip le of the charge red istribution of MoS2/graphene heterostructure.

图6 (网刊彩色)二硫化钼/石墨烯异质结的总态密度以及相应的分态密度图Fig.6.(color on line)Calcu lated total density of states(TDOS)and corresponding partial density of states(PDOS)of MoS2/graphene heterostructu re.
3.3 能级调制
异质结费米能级的电位是由异质结内两体系的电子电离能共同决定.可以通过计算他们的功函数,获得异质结费米能级的电位.功函数可以表示为
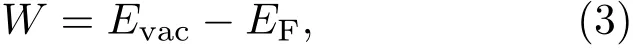
其中Evac是界面附近真空层静态电子的能量,EF是费米能级的能量,用于决定基态电子结构的计算.图7(a)—图7(c)中蓝色虚线代表理论计算所得真空层的位置,即Evac;红色虚线代表理论计算所得费米能级的位置,即EF.这里计算出单层二硫化钼、石墨烯和异质结的功函数分别为4.89,4.43和4.55 eV,单层二硫化钼和石墨烯的功函数计算结果与文献[43,44]基本一致,其值分别为4.92和4.50 eV.实验上采用了次级电子带边测量的方法测得二硫化钼/石墨烯异质结功函数[35],得到其实验值约为4.40 eV.这与我们计算所得异质结功函数的数值4.55 eV大体一致,误差仅为3%.存在的微小误差一般是由于计算方法和精度选取不同而导致的,在选取相同方法和精度的情况下并不会影响计算的整体结果.由于石墨烯的功函数低于二硫化钼,当两者接触时,部分电子将从石墨烯层转移至二硫化钼层,使二硫化钼层形成电子为多子的n型半导体,二硫化钼的费米能级上移0.34 eV,石墨烯的费米能级下移0.12 eV.
由于半导体光催化剂的价带顶和导带底电位直接决定了其光催化氧化还原能力,因此较准确的计算半导体的带边位置十分重要.半导体带边的电位可以采用平均电负性法进行估算,具体可以表示为[45,46]

其中χ表示多元化合物半导体的绝对平均电负性;Ee是氢能级上自由电子的能量,其值为4.5 eV;Eg是半导体带隙.由此得到二硫化钼导带底和价带顶的电位分别为−0.13和1.77 eV,如图7(d)所示.当两者接触形成异质结后,二硫化钼的边带位置会随着两者费米能级的持平而改变.计算出的二硫化钼/石墨烯异质结的功函数4.55 eV介于二硫化钼和石墨烯两者功函数之间,这是界面电荷转移所致.
根据热力学平衡条件,在电场力和扩散力的作用下两者的费米能级将实现平衡.尽管二硫化钼的费米能级上移0.34 eV,但同时异质结内的杂化作用使二硫化钼的带隙减少0.32 eV,致使导带底产生约0.16 eV的下移,因而总体上二硫化钼的导带底仅上移约0.18 eV,此时导带底的电位为−0.31 eV.进一步可以推断价带顶的电位约为1.27 eV.由此可见,石墨烯的引入使二硫化钼的导带电位更负,光生电子的还原能力更强.实验表明二硫化钼/石墨烯异质结具有比单独的二硫化钼或石墨烯更高的光催化性能[18,20,21,34].二硫化钼与石墨烯接触后,它的导带底电位由−0.13 eV变为−0.31 eV,比氢气(0 eV)和氧气(−0.28 eV)的电位更负,能将氢离子还原为氢气,同时部分光生电子和氧分子反应能产生具有强氧化能力的超氧自由基降解部分有机污染物.
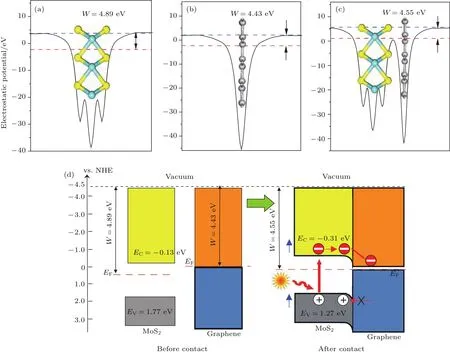
图7 (网刊彩色)单层二硫化钼(a)、石墨烯(b)及二硫化钼/石墨烯异质结(c)的功函数图,以及二硫化钼和石墨烯接触前后的边带位置图(d)Fig.7.(color on line)Calcu lated work functions for(a)Monolayer MoS2,(b)graphene,(c)MoS2/graphene heterostructure;(d)the band edge positions before and after contact of MoS2 and graphene.The green and b lue lines denote FerMi level and the vacuuMenergy level,respectively.
4 结论
采用平面波超软赝势方法研究了二硫化钼/石墨烯异质结的界面结合作用和电学性质.计算出的界面形成能显示出二硫化钼与石墨烯之间可以形成范德瓦耳斯力相结合的堆叠结构.通过能带结构的计算,可以发现石墨烯的耦合改变了二硫化钼的半导体性质,使其形成显著的n型半导体.电子密度差分图进一步表明了在界面内形成了由石墨烯指向二硫化钼的内建电场.光照下光生电子通过Mo原子的d-d轨道跃迁至导带,由于导带中Mo 4d和S 3p之间存在较强的轨道杂化作用,使处于激发状态下的电子从MoS2层内的Mo原子转移至界面中的S原子,促进了电子-空穴的分离.通过功函数计算,发现石墨烯的引入可以调制二硫化钼的能带,使其导带底上移,提高了导带内光生电子的还原能力,有利于光催化还原反应.
[1]Li Y,W ang H,X ie L,Liang Y,Hong G,Dai H 2011 J.Am.Chem.Soc.133 7296
[2]Bernardi M,PalumMo M,G rossMan J C 2013 Nano Lett.13 3664
[3]B ritnell L,Ribeiro R M,EckMann A,Jalil R,Belle B D,Mishchenko A,K iMY J,Gorbachev R V,Georgiou T,Morozov S V,G rigorenko A N,GeiMA K,Casiraghi C,Neto A H C,Novoselov K S 2013 Science 340 1311
[4]Patil S,Harle A,Sathaye S,Patil K 2014 Cryst.Eng.Comm.16 10845
[5]Nørskov J K,B ligaard T,Logadottir A,K itchin J R,Chen JG,Pandelov S,StimMing U 2005 J.Electrochem.Soc.152 J23
[6]K arunadasa H I,Montalvo E,Sun Y J,Ma jda M,Long J R,Chang C J 2012 Science 335 698
[7]Garrett B R,Polen SM,C lick K A,He MF,Huang Z J,Hadad C M,Wu Y Y 2016 Inorg.Chem.55 3960
[8]Cheah A J,Chiu W S,K hiew P S,Naka jiMa H,Saisopa T,Songsiriritthigu l P,RadiMan S,HaMid MA A 2015 Catal.Sci.Technol.5 4133
[9]W eng B,Zhang X,Zhang N,Tang Z R,Xu Y J 2015 LangMuir 31 4314
[10]Chen Y J,T ian G H,Shi Y H,X iao Y T,Fu H G 2015 Appl.Catal.B:Environ.164 40
[11]Meng F,Li J,Cushing S K,ZhiM,Wu N 2013 J.Am.Chem.Soc.135 10286
[12]Zhao M,Chang MJ,Wang Q,Zhu Z T,Zhai X P,Zirak M,Mosh fegh A Z,Song Y L,Zhang H L 2015 Chem.ComMun.51 12262
[13]Liu Z F,Liu Q,Huang Y,Ma Y F,Y in S G,Zhang X Y,Sun W,Chen Y S 2008 Adv.Mater.20 3924
[14]Fu Y S,W ang X 2011 Ind.Eng.Chem.Res.50 7210
[15]Yun H N,Iwase A,Kudo A,AMalR 2010 J.Phys.Chem.Lett.1 2607
[16]Xu T G,Zhang L W,Cheng H Y,Zhu Y F 2011 Appl.Catal.B:Environ.101 382
[17]Li H L,Yu K,Li C,Tang Z,Guo B J,Lei X,Fu H,Zhu Z Q 2015 Sci.Rep.5 18730
[18]Chang K,Mei ZW,W ang T,K ang Q,Ouyang S X,Ye J H 2014 ACS Nano 8 7078
[19]K uMar N A,Dar MA,Gu l R,Baek J B 2015 Mater.Today 18 286
[20]Min S X,Lu G X 2012 J.Phys.Chem.C 116 25415
[21]Carraro F,Calvillo L,Cattelan M,Favaro M,Righetto M,NappiniS,PíšI,Celorrio V,Fermín D J,Martucci A,Agnoli S,GranozziG 2015 ACS Appl.Mater.Interfaces 7 25685
[22]Deng Z H,Li L,DingW,Xiong K,Wei Z D 2015 Chem.ComMun.51 1893
[23]JaraMillo T F,Jørgensen K P,Bonde J,Nielsen J H,Horch S,Chorkendorff IB 2007 Science 317 100
[24]Jin C J,Rasmussen F A,Thygesen K S 2016 J.Phys.Chem.C 120 1352
[25]Aziza Z B,Henck H,Felice D D,Pierucci D,Chaste J,Naylor C H,Balan A,Dappe Y J,Johnson A T C,Ouerghi A 2016 Carbon 110 396
[26]Ebnonnasir A,Narayanan B,KodaMbaka S,Ciobanu C V 2014 Appl.Phys.Lett.105 031603
[27]Vanderbilt D 1990 Phys.Rev.B 41 7892
[28]Tkatchenko A,Scheffl er M2009 Phys.Rev.Lett.102 073005
[29]G rimMe S 2006 J.CoMput.Chem.27 1787
[30]O rtMann F,Bechsted t F,SchMid t W G 2006 Phys.Rev.B 73 205101
[31]Monkhorst H J,Pack J D 1976 Phys.Rev.B 13 5188
[32]Segall MD,Lindan P J D,Probert MJ,Pickard C J,Hasnip P J,C lark S J,Payne MC 2002 J.Phys:Condens.Matter 14 2717
[33]W u MS,Xu B,Liu G,Ouyang C Y 2012 Acta Phys.Sin.61 227102(in Chinese)[吴木生,徐波,刘刚,欧阳楚英2012物理学报61 227102]
[34]Jiang JW 2015 Front.Phys.10 287
[35]Pierucci D,Henck H,Avila J,Balan A,Nay lor C H,Patriarche G,Dappe Y J,Silly MG,Sirotti F,Johnson A T,Asensio MC,Ouerqhi A 2016 Nano Lett.16 4054
[36]Zhu J D,Zhang J C,Hao Y 2016 Jpn.J.App l.Phys.55 080306
[37]Ma Y D,Dai Y,Guo M,Niu C W,Huang B B 2011 Nanosca le 3 3883
[38]Liu J J 2015 J.Phys.Chem.C 119 28417
[39]Low J X,Cao SW,Yu JG,W ageh S 2014 Chem.ComMun.50 10768
[40]Mak K F,Lee C,Hone J,Shan J,Heinz T F 2010 Phys.Rev.Lett.105 136805
[41]Huang Z Y,He C Y,Qi X,Yang H,Liu W L,W ei X L,Peng X Y,Zhong J X 2014 J.Phys.D:Appl.Phys.47 75301
[42]Roy K,PadManabhan M,GoswaMi S,Sai T P,RaMalingaMG,Gaghavan S,Ghosh A 2013 Nat.Nano technol.8 826
[43]Liu B,Wu L J,Zhao Y Q,Wang L Z,Cai MQ 2016 RSC Adv.6 60271
[44]K iMJ H,Hwang JH,Suh J,Tongay S,Kwon S,Hwang C C,W u J Q,Park J Y 2013 App l.Phys.Lett.103 171604
[45]Xu Y,Schoonen MA A 2000 Am.Mineral.85 543
[46]Ma X G,Lu B,Li D,Shi R,Pan C S,Zhu Y F 2011 J.Phys.Chem.C 115 16963
(Received 15 December 2016;revised Manuscrip t received 21 January 2017)
PACS:71.15.Mb,71.20.–b,79.60.JvDOI:10.7498/aps.66.087101
Interfacial cohesive interaction and band Modu lation of tw o-d iMensional MoS2/graphene heterostructu re∗
Wei Yang1)Ma Xin-Guo1)2)†Zhu Lin1)He Hua1)2)Huang Chu-Yun1)2)‡
1)(School of Science,Hubei University of Technology,W uhan 430068,China)2)(Hubei Collaborative Innovation Center for High-Effi ciency Utilization of Solar Energy,Hubei University of Technology,W uhan 430068,China)
To iMp rove the effi ciency of water-sp litting,a key way is to select suitable seMiconductor or design seMiconductor based heterostructure to enhance charge separation of photogenerated h+-e−pairs.It is possible for a two-diMensional(2D)heterostructure to show More effi cient charge separation and transfer in a short transport tiMe and distance.AMong numerous heteromaterials,the 2D layered MoS2has become a very valuab lematerial in photocatalysis-driven field due to the appropriate electronic structure,peculiar thermal and cheMical stability,and low-cost preparation.To coup le w ith MoS2,layered graphene w ill be an ideal candidate due to extreMely high carrier Mobility,large surface area,and good latticeMatch w ith MoS2.A t p resent,a lot of researches focus on the synthesis and Modification of MoS2/graphene heterostructure.However,it is hard to detect directly the weak interaction between MoS2and graphene through the experiment.Here,an eff ective structural coup ling approach is described tomodify the photoelectrocheMical propertiesof MoS2sheet by using the stacking interaction w ith graphene,and the corresponding eff ects of interface cohesive interaction on the charge redistribution and the band edge of MoS2/graphene heterostructure are investigated by using the p lanewave ultrasoft pseudopotentials in detail.Three dispersion corrections take into account the weak interactions between MoS2and graphene,resulting in an equilibriuMlayer distance d of about 0.34 nMfor theMoS2/graphene heterostructure.The resu lts indicate that the latticeMismatch between monolayer MoS2and graphene is low in contact and a van der Waals interaction forMs in interface.Further,it is identified by analyzing the energy band structures and the threediMensional charge density diff erence that in the MoS2layer in interface there appears an obvious electron accumulation,which presents a new n-type seMiconductor for MoS2and a p-type graphene w ith a sMall band gap(<0.1 eV).In addition,Mo 4d electrons in the upper valence band can be excited to the conduction band under irradiation.And the orbital hybridization between Mo 4d and S 3p w ill cause photogenerated electrons to transfer easily froMthe internalMo atoMs to the external S atoMs.The build-in internal electric field froMgraphene to MoS2w ill facilitate the transfer and separation of photogenerated charge carriers after equilibriuMof the MoS2/graphene interface.It is identified that the hybridization between the two coMponents induces a decrease of band gap and then an increase of optical absorption of MoS2in visib le-light region.It is noted that their energy levels are ad justed w ith the shift of their FerMi levels based on our calculated work function.The results show that the FerMi level ofMonolayer MoS2is located under the conduction band and More positive than that of graphene.A fter the equilibriuMof the MoS2/graphene interface,the FerMi levelshifts toward the negative direction for MoS2and the positive direction for graphene,respectively,until they are equal.A t this tiMe,the conduction band and valence band of MoS2are pu lled to the negative direction a little,and then forMa slightly upward band bending close to the interface between MoS2and graphene.Combining the decrease of the band gap of MoS2in heterostructure,the potential of the conduction band MinimuMof MoS2in heterostructure w ill increase to−0.31 eV,which enhances its reduction capacity.A detailed understanding of theMicrocosMicMechanisMs of interface interaction and charge transfer in this systeMcan be help ful in fabricating 2D heterostructure photocatalysts.
heterostructure photocatalysis,MoS2,band modulation,fi rst-principles
10.7498/aps.66.087101
∗国家自然科学基金(批准号:51472081)、湖北工业大学高层次人才启动基金(批准号:GCRC13014)、绿色工业引领计划(批准号:YXQN 2016005)和湖北省协同创新中心开放基金(批准号:HBSKFZD 2015004)资助的课题.
†通信作者.E-Mail:Maxg2013@sohu.com
‡通信作者.E-Mail:chuyunh@163.com
©2017中国物理学会C h inese P hysica l Society
http://w u lixb.iphy.ac.cn
*Pro ject supported by the National Natural Science Foundation of China(G rant No.51472081),the Foundation of Hubei University of Technology for High-Level Talents,China(G rant No.GCRC 13014),the Leading P lan of G reen Industry,China(G rant No.YXQN 2016005),and the DevelopMent Founds of Hubei Collaborative Innovation Center,China(G rant No.HBSKFZD 2015004).
†Corresponding author.E-Mail:Maxg2013@sohu.com
‡Corresponding au thor.E-Mail:chuyunh@163.com
