以石墨烯为电极的有机噻吩分子整流器的设计及电输运特性研究∗
2017-08-09俎凤霞张盼盼熊伦殷勇刘敏敏高国营
俎凤霞 张盼盼 熊伦†殷勇 刘敏敏 高国营
1)(武汉工程大学理学院,武汉 430205)2)(华中科技大学物理学院,武汉 430074)
以石墨烯为电极的有机噻吩分子整流器的设计及电输运特性研究∗
俎凤霞1)张盼盼1)熊伦1)†殷勇1)刘敏敏1)高国营2)
1)(武汉工程大学理学院,武汉 430205)2)(华中科技大学物理学院,武汉 430074)
(2016年10月29日收到;2017年1月20日收到修改稿)
传统硅基半导体器件受到了量子尺寸效应的限制,发展分子电子学器件有可能解决这一难题.本文提出了由石墨烯电极和有机噻吩分子相结合构造分子器件的思想,建构了“石墨烯-噻吩分子-石墨烯”结构的分子器件,并运用非平衡态格林函数结合密度泛函理论的方法研究了其电输运特性.系统地分析了电子给体“氨基”和电子受体“硝基”两种取代基的位置对有机噻吩分子电输运的影响.计算表明,有机噻吩二聚物被“氨基”和“硝基”取代后会产生明显的负微分电阻效应和整流效应.进一步对产生这些效应的物理机制进行分析,发现氨基的位置可以调整负微分电阻的强弱,硝基的位置可以改变整流的方向.
分子电子学,非平衡格林函数,石墨烯,噻吩
1 引 言
近年来,随着科技的进步,基于单分子级别的分子电子学器件,比如分子开关、分子二极管、分子场效应管等,成为当前的研究热点之一[1−4].这些具有逻辑功能和运算功能的分子电子学研究成果有望推动计算机技术突破传统硅器件的限制,制作出高性能小体积的电子器件.其中,分子整流器发挥了至关重要的作用,一方面因为它是第一个单分子器件,另一方面因为整流器是电路中最简单的而且是不可缺少的单元.1974年,Aviram和Ratner[5]首次从理论上提出分子整流器的概念,这是关于分子电子器件的最早研究.此后,关于分子整流器的理论和实验报道更是层出不穷[6−8].2016年4月,IT之家网站报道了《自然化学》期刊的一项关于分子电子学的科研成果[9],研究人员通过对DNA分子的双螺旋结构内部插入两个小的Coralyne分子,将其连接到两端的金电极形成分子器件,对其施加偏压时发现该分子器件具有二极管的功能.这款新型的二极管的长度只有11个碱基对,被认为是全球尺寸最小的二极管.由于基因科学的突破,使精确设计DNA成为可能,这项研究也使得分子电子学器件投入到实际的生产应用成为可能.在分子整流器的构造中,分子的选择是一个关键因素.具有退局域化π电子的有机噻吩分子由于其良好的电和光属性以及低的共振能和更好的稳定性被认为是构造分子器件的理想建构原材料之一[10−12].2002年,Zhitenev等[13]采用一种新的实验方法,成功制备了“金属-噻吩分子-金属”分子结,研究了源-漏电压、门电压调控的电输运属性,这是第一个基于单分子的分子场效应管,也实现了真正意义上的“单个”分子器件.2013年,杨英[14]对乙基噻吩分子器件的电子运输性质进行了研究,得出影响分子器件的伏安特性的两个因素分别为金属表面和分子之间的相互作用能以及分子本身的电子结构.还有更多的实验和理论研究利用有机噻吩分子形成分子结并调查了其量子输运属性[15−17].然而,这些研究的绝大多数都是使用“金”作为分子器件的电极.本文另辟蹊径,使用“石墨烯”作电极构造了一系列基于噻吩的分子器件.与金属电极相比,“石墨烯”作电极能有效地避免了金属电极易氧化以及有机分子与无机金属电极界面不稳定等问题.首先,本文设计了一系列基于噻吩二聚物衍生物的整流器件,主要是将噻吩二聚物中的两个氢原子分别被电子给体氨基和电子受体硝基取代,形成非对称结构.其次,运用基于非平衡格林函数结合密度泛函的第一性原理方法对上述分子器件执行了电输运计算.计算结果表明这些分子器件都具有整流效应及负微分电阻效应.进一步,从分子电子结构、输运谱及分子投影本征态等几个方面分析了其电输运特性的物理机制,期望本文的研究结果能为今后分子电子器件的设计提供基本的参考.
2 建构模型及计算方法
本文选择有机噻吩二聚物分子(thiophene dimer,2T)作为研究对象,构筑了噻吩原型(2T-0)以及四种被电子给体“氨基”(—NH2)和电子受体“硝基”(—NO2)取代后的噻吩二聚物分子衍生物整流器,它们分别是:2T-14,2T-13,2T-24和2T-23,如图1所示.其中,2T-14结构中,—NH2和—NO2分别取代分子中的1,4位置的两个氢原子;2T-13结构中取代的是1,3位置;2T-24结构取代2,4位置;2T-23结构则是取代2,3位置.
不同于以前的研究,本文选用石墨烯为电极,上述四种结构的分子为中心散射区组成不同结构的器件,结构如图2所示.所有器件都是由左电极、中心散射区和右电极三部分组成.左右石墨烯电极选用的是经过裁剪的锯齿型石墨烯纳米带,其边缘碳原子中的未饱和键都被氢原子钝化.石墨烯可以通过裁剪获得结构不同的纳米带,比如常见的锯齿型(zigzag)石墨烯纳米带和扶手椅型(armchair)的石墨烯纳米带.为什么选择锯齿型石墨烯纳米带作电极?第一性原理计算预测扶手椅型的石墨烯纳米带的电子结构显示其具有半导体属性,且带隙随纳米带的宽度变化[18,19],而锯齿型石墨烯纳米带则呈现金属性,适合作为分子器件的电极材料.在石墨烯纳米带电极的构造中,左右电极在输运的z方向是半无限长的,在与输运垂直的x和y方向都加了足够的真空层,以抑制相邻分子间的相互作用.中心散射区包含噻吩衍生物分子和石墨烯纳米带电极的表面层,以屏蔽分子和电极的相互影响.

图1 (网刊彩色)噻吩二聚物分子原型2T-0以及四种被“—NH2”和 “—NO2”取代后的噻吩分子衍生物2T-14,2T-13,2T-24,2T-23Fig.1.(color online)Thiophene dimer 2T-0 and its derivatives,such as 2T-14,2T-13,2T-24 and 2T-23 substituted by—NH2and—NO2.
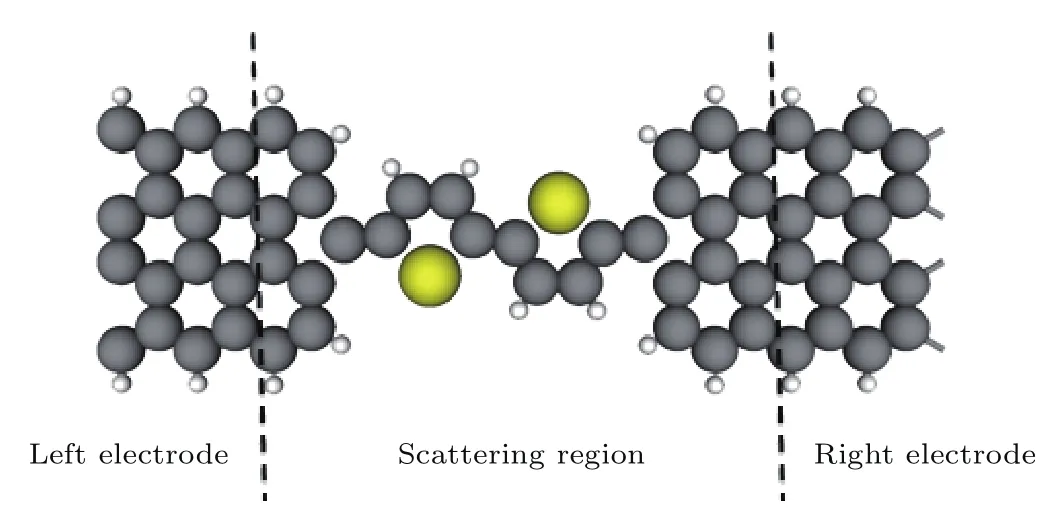
图2 (网刊彩色)分子器件的结构,中心区2T-0分子可以替换为2T-14,2T-13,2T-24和2T-23Fig.2.(color online)The structure of molecular devices,and the central molecules can be 2T-0,2T-14,2T-13,2T-24 and 2T-23.
本文对器件的建构、电子结构以及输运性质是利用ATK软件包[20−22]进行研究的.首先我们对中心散射区的结构进行了优化,并进行自洽计算获得初始电子密度.在密度泛函的参数设置中,基矢是双链极化基(double zeta with polarization)、交换关联泛函为广义梯度近似下的Perdew-Burke-Ernzerhof泛函、截止能(the mesh cut-o ffenergy)为150 Ry,k点取样模板用(1,1,100)及总能量收敛标准10−5,选取这些参数可满足准确的自洽收敛.
3 计算结果
3.1噻吩二聚物原型的电输运特性
首先我们研究了噻吩二聚物原型2T-0分子器件的电输运,作为后面噻吩二聚物衍生物的研究参考.通过对分子器件两端施加偏压,利用下面的公式得到了噻吩分子器件的电流-电压(I-V)曲线,如图3(a)所示.

其中µL,R分别是左右电极的化学势,T(E,Vb)是在能量为E、偏压为Vb时的输运系数.
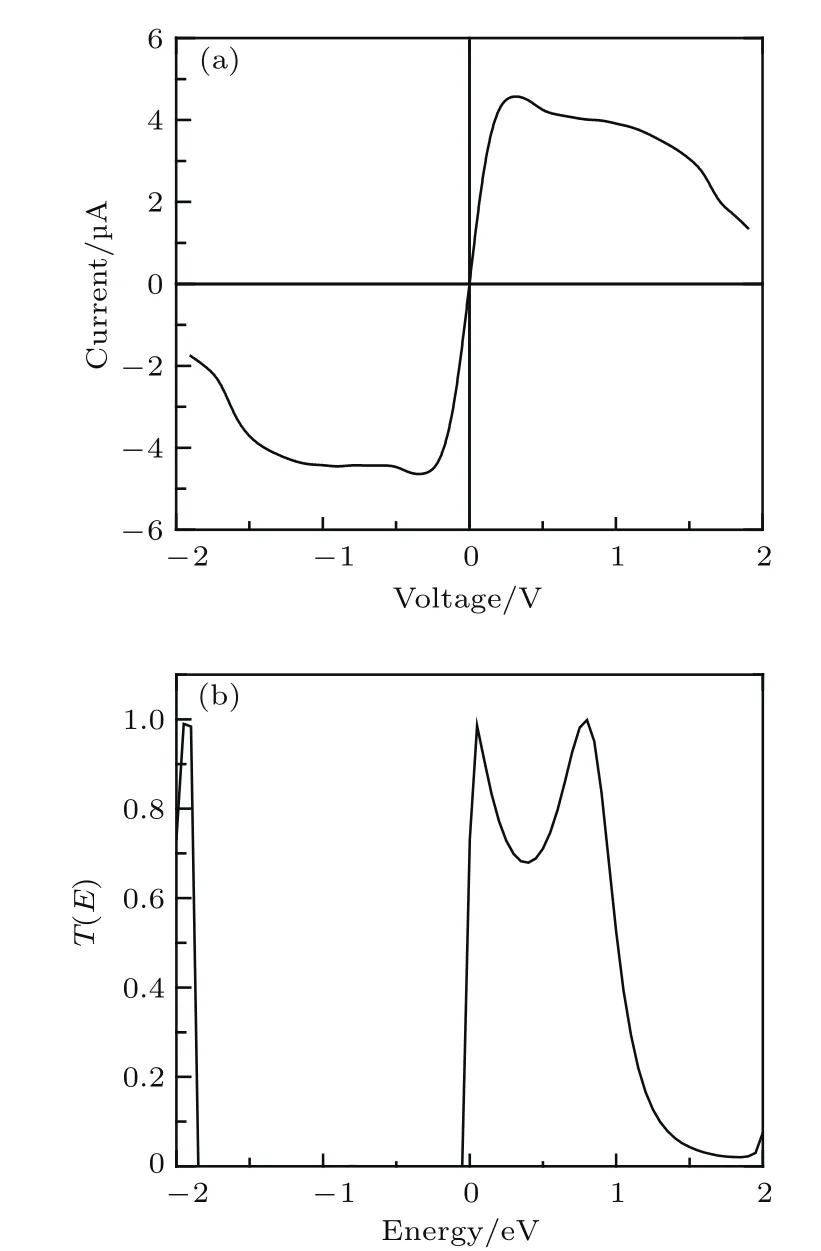
图3 噻吩二聚物原型2T-0的电输运属性 (a)电流-电压曲线;(b)零偏压下的输运谱Fig.3.The transport properties of 2T-0:(a)The current-voltage curve;(b)the transmission coefficient at zero bias.
由图3(a)可知,正负偏压下的电流-电压曲线基本呈现对称分布.在低的偏压范围内,电流随电压呈线性增长;当偏压增加到0.3 V时,电流达到一个峰值.之后,随着偏压的增大,电流开始非线性下降,呈现明显的负微分电阻效应.考虑到电流与输运系数相对应,所以我们也计算了2T-0分子器件在零偏压下的输运谱,如图3(b)所示.在费米面附近的负能量区域,输运系数几乎为零,而在费米面以上的正能量区域,出现了一个比较宽的输运峰.这个输运谱是分子器件电流的主要来源.由于它位于费米面上方,我们预测这个输运峰主要来源于费米面以上的分子能级.
3.2噻吩二聚物衍生物的电输运特性
在上述噻吩二聚物原型分子器件2T-0的电输运基础上,我们进一步计算了被电子给体“—NH2”和电子受体“—NO2”取代后的噻吩二聚物分子衍生物的电输运特性,图4给出了2T-14,2T-13,2T-24和2T-23四种结构分子器件的电流-电压曲线,它们呈现以下特征.1)非对称性.每条曲线在正偏压时的电流都不同于相应的负偏压电流,导致了明显的整流行为,这与上述原型器件2T-0的对称性输运特性不同.进一步,为了详细说明电流-电压曲线的非对称性,我们计算了各体系的整流比,定义为R(V)=|I(+V)/I(−V)|,其中,I(+V)与I(−V)分别表示相应的正负偏压下的电流.R(V)=1说明没有整流,R(V)>1说明正偏压的电流大于负偏压,反之,R(V)<1则表示负偏压的电流大于正偏压电流.由于更高偏压时电流不稳定,故图4只显示了低偏压下的整流比.由图4可知,2T-14和2T-13结构的分子器件,正偏压下的电流小于对应负偏压的电流,呈现了负整流效应;而2T-24和2T-23结构,总体上表现为正整流行为.需要指出,这四个被“—NH2”和“—NO2”取代后的噻吩二聚物分子衍生物结合石墨烯电极形成的器件,整流比不是很可观.然而,对比过去的研究使用噻吩二聚物分子原型和金电极形成的器件,这四个器件的整流效果更加明显[16].2)非线性特征.所有结构的电流随着偏压的增加非线性变化,显示了比原型器件2T-0更明显的负微分电阻峰.3)取代基位置不同,输运不同.结构2T-14和2T-13的—NO2位置相同、—NH2不同,它们的电流-电压曲线的形貌相似,仅仅2T-13电流比对应偏压下2T-14的电流大,负微分电阻峰更尖锐.而2T-24和2T-13电流-电压曲线似乎关于正负偏压镜像分布(2T-24正偏压下电流与2T-13负偏压相似,2T-24负偏压电流与2T-13正偏压相似).
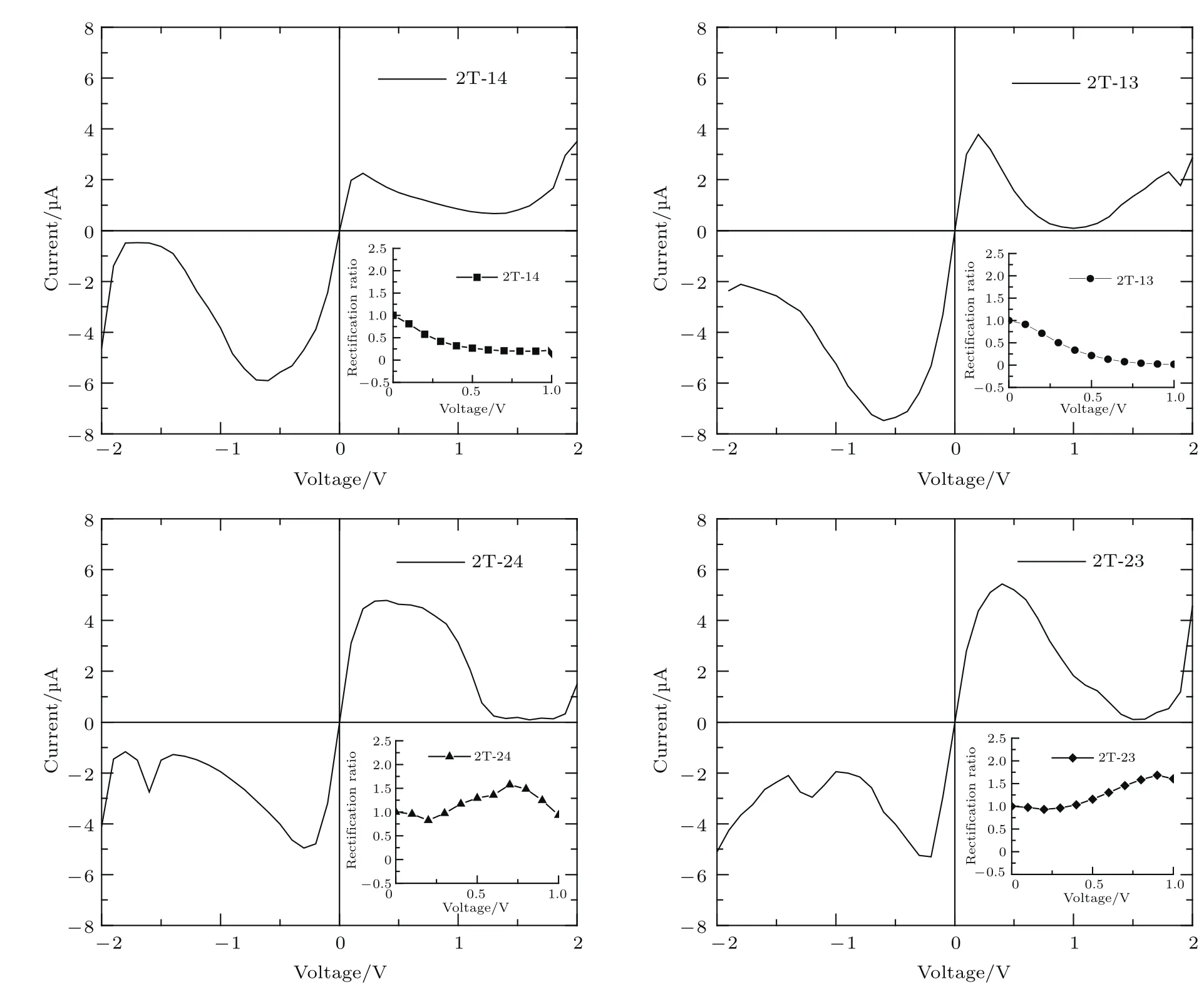
图4 噻吩二聚物分子衍生物2T-14,2T-13,2T-24和2T-23四种结构的电流-电压特性和整流比(内插图)Fig.4.The current and recti fi cation ratios(R)as a function of applied bias voltage for the thiophene derivatives 2T-14,2T-13,2T-24,and 2T-23,respectively.
4 结果分析与讨论
4.1零偏压下的输运谱
为了解释上述输运特征,图5只给出了它们在零偏压下的输运谱.与原型器件2T-0的输运谱相似,所有四个结构分子器件的输运谱都位于费米能级上方.而且无论是对原型器件2T-0,还是四个衍生物分子器件,紧邻费米面的这个宽的主输运峰的形貌十分相似:主输运峰的顶端被劈裂为两个子峰.其中一个靠近费米面,在所有器件中,这个峰都位于+0.05 eV能量位置,不受取代基影响.另一子峰,虽然在原型器件和所有衍生物器件中都有出现,可是它的位置、高度对不同结构的分子器件是不同的.图3(b)所示的器件原型的主峰最宽,且两个劈裂峰的峰值相当,输运谱被拓宽,导致图3(a)中负微分电阻随电压的增大呈缓慢下降的趋势.相比较,分子衍生物2T-14,2T-13,2T-24和2T-23四种结构的主峰变窄,使得负微分电阻效应更明显.其中,2T-14的输运峰最低,对应的电流也是所有器件中最小的.
4.2分子投影能谱及本征态
分子本身的电子结构是决定分子器件电输运的重要因素,因此,为了定性分析取代基—NH2和—NO2对输运性质的影响,我们给出了分子投影能谱以及对应的分子投影自洽哈密顿态(molecular projected self-consistent Hamiltonian states,MPSHs).考虑到对输运起作用的主要是费米面附近的能级,所以图6只给出了最高分子占有轨道(highest occupied molecular orbital,HOMO),最低未占有分子轨道(lowest unoccupied molecular orbital,LUMO),以及次低未占有分子轨道(LUMO+1)能级.

图5 噻吩二聚物分子衍生物2T-14,2T-13,2T-24和2T-23四种结构在零偏压下的输运谱(费米能级被设置为0)Fig.5.The transmission coefficients of the thiophene derivatives at zero bias for 2T-14,2T-13,2T-24,and 2T-23,respectively.
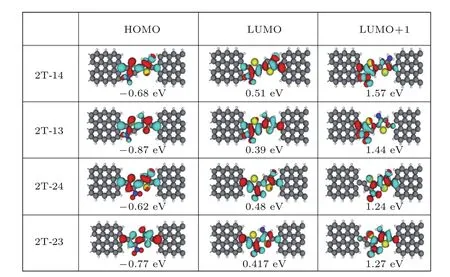
图6 (网刊彩色)噻吩二聚物分子衍生物2T-14,2T-13,2T-24和2T-23四种结构在零偏压下的能级以及对应的MPSHsFig.6.(color online)The energy spectra and the MPSHs at zero bias for 2T-14,2T-13,2T-24,and 2T-23,respectively.
所有四个结构分子器件的HOMO能级对应的MPSH态在—NH2上都有分布,在—NO2上几乎没有(除了2T-14和2T-13中有极少的态分布到—NO2的O原子上);与之相反,LUMO及LUMO+1能级对应的MPSH态在—NO2上都有分布,在—NH2上分布较少(特别是LUMO+1态在—NH2上分布几乎为零).说明—NH2主要是对HOMO能级有贡献,而—NO2主要贡献于LUMO及LUMO+1,这与—NH2是电子给体、—NO2是电子受体的本质相一致.进一步,我们发现2T-14和2T-24分子结构的HOMO,LUMO能级都比较高;2T-13和2T-23分子结构的HOMO,LUMO能级则都比较低,似乎整体向能量负向漂移.2T-14和2T-24结构—NH2的取代位置相同,都处在4位置;而2T-13和2T-23结构—NH2都取代3位置,可见—NH2的取代位置会直接影响HOMO,LUMO能级的移动方向,处在最外侧的4位置对应的能级偏高.对照图5,四个分子器件的主要的输运峰都来源于费米能级上方的LUMO能级.2T-14和2T-24结构(—NH2在4位)的LUMO能级远离费米能级,这两个结构的主输运峰比较宽,对应的负微分电阻变化较平缓;2T-13和2T-23结构(—NH2在3位)的LUMO能级接近费米能级,这两个结构的主输运峰比较窄,对应的负微分电阻变化较陡.这表明—NH2位置会影响主输运峰的宽度,处在分子内侧的3位置,负微分电阻效应更明显.
4.3不同偏压下的输运谱
为了进一步理解上述I-V曲线中的整流效应和负微分电阻效应背后的物理机制,我们分析了不同偏压下的输运谱,考虑到2T-14和2T-13结构的输运特性类似,即在低偏压范围内负偏压的电流大于对应的正偏压电流;而2T-24和2T-23结构的输运特性类似,即在低偏压范围内正偏压的电流大于对应的负偏压电流,进而使得这两类结构的整流方向正好相反.为了定性分析这两者的区别,下面只选择2T-13和2T-24分子器件的输运谱为例来分析.图7(a)和图7(b)给出了这两个器件在6个特殊的偏压下的输运谱.由于电流是由输运系数
T(E,Vb)对偏压窗口[−Vb/2,+Vb/2]范围的积分获得的,所以我们只分析偏压窗口内(两条蓝色竖线包含的区域)的输运谱变化.
1)负微分电阻效应机制.图7(a)和图7(b)的共同特征是费米面附近的主要输运峰随着偏压的增大向能量的正方向漂移,导致正负偏压下都出现了负微分电阻效应.鉴于正负偏压下的负微分电阻的机理类似,下面我们只分析正偏压下的负微分电阻效应.先来分析2T-13分子器件,图7(a)显示施加偏压后,偏压窗口加宽,输运谱开始进入了偏压窗口,导致电流也从零逐渐增大,当偏压达到+0.2 V时,电流达到极值.然而,当偏压继续增大时,输运谱的峰值迅速降低,而且向正能量的方向漂移,逐渐远离了偏压窗口(见+0.6 V和+1.0 V),电流随之陡然下降,形成了图4电流-电压曲线显示的明显的负微分电阻效应.相比较,图7(b)显示2T-24分子器件的输运谱在+0.3 V(对应电流最大)之后也远离了偏压窗口,但偏压窗口内的输运峰仍较大(见+0.8V),电流电压曲线在+0.3 V到+0.8 V范围内下降比较缓慢,负微分电阻效应较弱,直到高偏压时才明显下降.总之,2T-14和2T-24结构的分子器件,—NH2都在4位置,负微分电阻随偏压的增加变化缓慢;而2T-13和2T-23结构,—NH2都处在3位置,负微分电阻峰更尖锐,负微分效应更显著.所以,—NH2的位置可以调整负微分电阻的强弱.
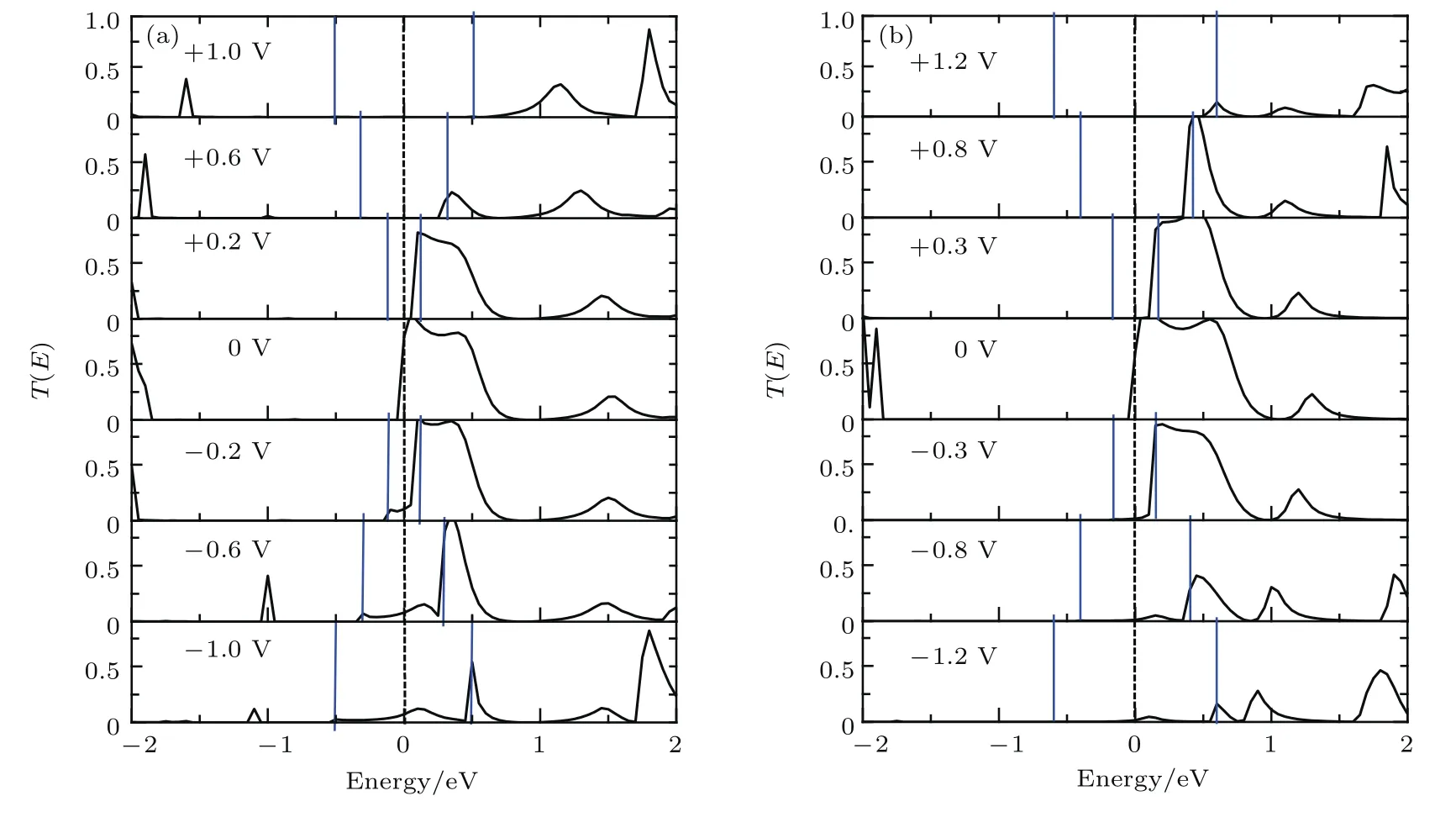
图7 (网刊彩色)噻吩二聚物分子衍生物(a)2T-13和(b)2T-24分子器件在6个特殊偏压下的输运系数,费米能级被设为零(虚线表示),两条竖线(蓝色)区域为偏压窗口Fig.7.(color online)The transmission coefficients of the thiophene derivatives at six special bias for(a)2T-13 and(b)2T-24.The average Fermi level is set to zero(dash line),and the region between two blue lines is the bias window.
2)整流效应机理.尽管图7(a)和图7(b)显示费米面附近的主要输运峰随着偏压的增大向正能量方向漂移,远离偏压窗口.然而,对于2T-13分子器件,图7(a)显示在负偏压时,偏压窗口内仍保留了部分输运谱,远大于正偏压时偏压窗口的输运谱,导致了负向的整流效应.2T-24分子器件则相反,图7(b)表明,在低的偏压范围内,负偏压时偏压窗口内的输运峰低于相应正偏压的输运峰,结果正偏压的电流大于相应负偏压电流,导致了正向的整流行为.综合来看,2T-14和2T-13结构的分子器件,—NO2都在1位置,呈现了负整流效应;而2T-24和2T-23结构,—NO2都处在2位置,会产生正整流行为.所以,—NO2的位置可以改变整流的方向.
5 结 论
本文提出了由石墨烯电极和有机噻吩分子相结合构造分子器件的思想,并运用非平衡态格林函数结合密度泛函理论的方法研究其电输运及特性,系统地分析了“—NH2”和“—NO2”取代基的位置对有机噻吩分子的电输运影响.计算表明,有机噻吩二聚物被“—NH2”和“—NO2”取代后会产生明显的负微分电阻效应和整流效应.进一步,我们通过计算分子投影自洽哈密顿态和输运谱分析了这些效应背后的物理机制,得出结论:氨基的位置可以调整负微分电阻的强弱,当它处在中间的位置时,负微分效应更显著;硝基的位置可以改变整流的方向,它处在中间位置,会产生正整流行为,在分子的边缘位置会导致负整流行为.
感谢华中科技大学物理学院姚凯伦教授为本研究指明了方向.
[1]Reed M A,Zhou C,Muller C J,Burgin T P,Tour J M 1997Science278 252
[2]Seminario J M,Zacarias A G,Tour J M 1999J.Am.Chem.Soc.121 411
[3]Zu F X,Liu Z L,Yao K L,Gao G Y,Fu H H,Zhu S C,Ni Y,Peng L 2014Sci.Rep.4 4838
[4]Jalili S,Ra fi i-Tabar H 2005Phys.Rev.B71 165410
[5]Aviram A,Ratner M A 1974Chem.Phys.Lett.29 277
[6]Taylor J,Brandbyge M,Stokbro K 2002Phys.Rev.Lett.89 138301
[7]Wang B,Zhou Y S,Ding X L 2006J.Phys.Chem.B110 24505
[8]Tongay S,Lemaitre M,Miao X,Gila B,Appleton,B R,Hebard A F 2012Phys.Rev.X2 011002
[9]Guo C L,Wang K,Zerah-Harush E,Hamill J,Wang B,Dubi Y,Xu B Q 2016Nat.Chem.8 484
[10]Yamada R,Hiroaki K,Noutoshi T,Tanaka S,Tada H 2008Nano Lett.8 1237
[11]Fichou D 2000J.Mater.Chem.10 571
[12]McCreery R L,Yan H J,Bergren A J 2013Phys.Chem.Chem.Phys.15 1065
[13]Zhitenev N B,Meng H,Bao Z 2002Phys.Rev.Lett.88 226801
[14]Yang Y 2013M.S.Thesis(Chengdu:Southwest Jiaotong University)(in Chinese)[杨英 2013硕士学位论文(成都:西南交通大学)]
[15]Zu F X,Liu Z L,Yao K L,Fu H H,Gao G Y,Yao W 2013J.Chem.Phys.138 154707
[16]Bao Q L,Lu Z S,Li J,Loh K P,Li C M 2009J.Phys.Chem.C113 12530
[17]Zhou Y X,Jiang F,Chen H,Note R,Mizuseki H,Kawazoe Y 2007Phys.Rev.B75 245407
[18]Barone V,Hod O,Scuseria G E 2006Nano Lett.6 2748
[19]Han M Y,Özyilmaz B,Zhang Y,Kim P 2007Phys.Rev.Lett.98 206805
[20]Taylor J,Guo H,Wang J 2001Phys.Rev.B63 245407
[21]Brandbyge M,Mozos J L,Ordejón P,Taylor J,Stokbro K 2002Phys.Rev.B65 165401
[22]Soler J M,Artacho E,Gale J D,García A,Unquera J,Ordejón P,Sánchez-Portal D 2002J.Phys.Condens.Matter14 2745
PACS:85.65.+h,73.63.–b,61.46.–w,72.80.VpDOI:10.7498/aps.66.098501
Design and electronic transport properties of organic thiophene molecular recti fi er with the graphene electrodes∗
Zu Feng-Xia1)Zhang Pan-Pan1)Xiong Lun1)†Yin Yong1)Liu Min-Min1)Gao Guo-Ying2)
1)(School of Science,Wuhan Institute of Technology,Wuhan 430205,China)2)(School of Physics,Huazhong University of Science and Technology,Wuhan 430074,China)
29 October 2016;revised manuscript
20 January 2017)
Molecular electronics o ff ers new possibilities for continually miniaturizing the electronic devices beyond the limits of standard silicon-based technologies.There have been signi fi cant experimental and theoretical e ff orts to build thiophene molecular junctions and study their quantum transport properties.However,in most of these studies Au is used as lead material.It is well known that the fabrication of the traditional molecular device is now hindered by technological difficulties such as the oxidation of metallic contacts,and the interface instability between the organic molecule and the inorganic metallic electrodes.In this paper,we use the graphene electrodes to construct a series of thiophene-based devices.The graphene electrodes proposed in this paper are able to avoid such problems.Moreover,the stability of graphene electrodes at room temperature paves the way to studying the electron transport through a single molecule under the ambient conditions.Firstly,we design a series of molecular rectifying devices based on thiophene dimer and its derivatives,in which the hydrogen atom on the thiophene monomer is substituted with a representative electrondonating group(—NH2)and electron-withdrawing group(—NO2).Secondly,we investigate systematically the electronic transport properties through these molecular junctions by performing the fi rst principles calculations based on density functional theory and nonequilibrium Green’s function.The calculated results show that these thiophene molecular devices substituted with—NH2and—NO2possess the rectifying behavior and negative di ff erential resistance properties.Furthermore,we also fi nd that the position of substituent group—NH2or—NO2has a major in fl uence on the electronic transport properties.In order to explore the physical mechanism behind these transport properties,the electronic structures of the molecules,the transmission spectrum,and the molecular projected eigenstates are analyzed.The results reveal that the position of NH2can adjust the intensity of the negative di ff erential resistance.When the NH2group is close to the molecular end,the negative di ff erential resistance behavior in this molecular device is more prominent than in other molecules.In addition,the position of NO2can change the direction of the recti fi cation.When the NO2group is close to the molecular end,the current in negative bias is larger than in positive bias,resulting in a negative recti fi cation.In contrast,when the NO2group is close to the molecular centre,a positive recti fi cation occurs.Our results can provide a worthy complement to thiophene molecular experiment,and also has a guiding signi fi cance for designing other molecular electronic devices.
molecular electronics,nonequilibrium Green’s function,graphene,thiophene
10.7498/aps.66.098501
∗湖北省教育厅科学研究计划(批准号:Q20151510)和武汉工程大学科学研究基金(批准号:K201477)资助的课题.
†通信作者.E-mail:13971624916@163.com
*Project supported by the Scienti fi c Research Foundation of Education Bureau of Hubei Province,China(Grant No.Q20151510)and the Scienti fi c Research Foundation of Wuhan Institute of Technology,China(Grant No.K201477).
†Corresponding author.E-mail:13971624916@163.com
