外源DNA在哺乳动物细胞中不依赖于启动子的转录和剪接现象
2017-08-07徐文杨华瑜郑永昌
徐文杨华瑜郑永昌
(1. 清华大学生命科学学院,北京 100084;2. 中国医学科学院北京协和医院肝脏外科,北京 100730)
外源DNA在哺乳动物细胞中不依赖于启动子的转录和剪接现象
徐文1杨华瑜2郑永昌2
(1. 清华大学生命科学学院,北京 100084;2. 中国医学科学院北京协和医院肝脏外科,北京 100730)
外源性导入哺乳动物细胞的双链DNA会经历重组、重排及其他复杂的分子生化反应。极少数DNA分子会随机整合到宿主基因组,即发生水平基因转移。然而,目前还不清楚这些外源性DNA分子在不含有特殊启动子的情况下是否能被细胞的转录机器识别并起始转录。在构建环状RNA(circRNA)表达体系的过程中,发现了外源性DNA分子能经历不依赖启动子的转录,而且转录本会发生符合“GU-AG”法则的RNA剪接进而产生类似于环状RNA的异常剪接分子。进一步研究表明,外源DNA在哺乳动物细胞中不依赖于启动子的转录和剪接现象是保守的。揭示了外源DNA在哺乳动物细胞中的一个新的命运途径,该发现对研究外源性DNA对宿主细胞的影响具有重要意义及应用价值。
不依赖启动子的转录;RNA剪接;circRNA;DNA转染;选择性剪接
早期研究发现,导入真核细胞的外源DNA分子可以发生重组、重排及其他复杂的分子生化反应,可能形成长达几百kb的头尾串联结构[1-7]。在哺乳动物卵子和受精卵中微注射的DNA也能通过这种方式形成类似的高分子化合物结构[8,9]。然而,对于这些DNA能否在缺乏功能性启动子下进行转录及其后转录本的剪接缺乏尚不清楚。
根据报道,一类称为coligo(Circular oligonucleotides,环状寡核苷酸小体)的单链环状DNA导入哺乳细胞后能被RNA聚合酶III识别并进行不依赖于启动子转录,产生功能性RNA分子[10]。其后的研究表明,coligo的有效转录也需要相关序列和结构的参与[11]。通过转录组测序发现导入哺乳动物细胞的无启动子DNA上存在部分区域的异常转录峰,具体机制尚不明确,也可能是在转录测序过程中存在DNA污染[12]。
我们在研究环状RNA的生成过程中偶然发现了不依赖于启动子的转录以及其后转录本的剪接现象。circRNA是近年发现的一类共价闭合的环状非编码RNA分子,其在细胞中的检测主要使用反向引物对进行RT-PCR[13-15]。本研究以cirRNA在人细胞中的过表达为出发点,借助RT-PCR和Northern blot等技术探究不依赖于启动子转录和剪接现象,并对转录本的性质作深入分析。
1 材料与方法
1.1 材料
1.1.1 细胞株 HEK 293T细胞和NIH 3T3细胞来自于ATCC。
1.1.2 主要试剂 pGEMT、pGL和pcDNA3.1载体分别购自Promega和Invitrogen生物公司。KOD-plus-突变试剂盒购自TOYOBO。细胞转染试剂Lipofectamine LTX、RNA提取试剂Trizol、反转录试剂盒SuperScript III First-Strand Synthesis system、荧光定量PCR试剂盒SYBR Green以及ULTRAhyb-Oligo杂交液均购自Life Technologies。引物合成和测序委托金唯智生物公司。DMEM培养基、胎牛血清和抗生素为Gibco公司产品。所有酶制剂均来自于NEB。
1.2 方法
1.2.1 基因片段克隆 根据从NCBI网站获得的不同基因片段序列信息以及文献报道,设计不同的引物对直接以少量细胞为模板进行PCR扩增[15]。所用引物对分别为zkscan1:5′-AACCAGTCTACATCACAATGTCTAA-3′,5′-AACCTAGAAAAGACTGAT-GCTATAGT-3′;ZNF608:5′-AAATGGGCTTTGTGTGTG-3′,5′-TCCTGCCCTTACTCCTCT-3′;CAMSAP1:5′-CGTCTTTA-GTGGGACAGCCGTT-3′;5′-AGCCAGCAGATGGAGCACC-3′;HIPK3:5′-GAAAGTCATTAACTTGTCTTTGTAATAT-3′;5′-GACCTAAAGAGTCACGGGAGC-3′;clec2d:5′-AGCTATCATCAGATTCACACCTCA-3′;5′-TCACATGACACTGCCTTTCATC-3′。PCR反应条件均参照Q5 DNA聚合酶说明书进行。
1.2.2 载体构建 上述片段按照常规TA克隆方法得到pGEMT系列载体。对于插入到pcDNA3.1的序列片段,使用5′端加入酶切位点的引物对PCR得到,引物对分别为pCDNA3.1_zkscan1:5′-ACTGggtacc-AACCAGTCTACATCACAATGTCTAA-3′,5′-ACTGgcg gccgcAACCTAGAAAAGACTGATGCTATAGT-3′;400-1882:5′-ATCGgatatcAAAGTGCTGAGATTACAGGCGT-3′,5′-ATCGgcggccgcTAATTTTCATATTTTTAGTAGAGACAAGG-3′。Zkscan1突变按照KOD-plus-突变试剂盒的说明进行,使用的突变引物对为:5′-CAAAGAAGCAAGGTTTCATTTAGG-3′和5′-CTCGTGACTGTAAGAGGC-3′。
1.2.3 细胞转染、总RNA提取与反转录 细胞使用Lipofectamine LTX脂质体转染试剂按常规方法转染,转染前细胞密度在70%左右,六孔板每孔转染的质粒为2 μg。转染24 h后,使用Trizol提取细胞总RNA,并使用DNase I处理去除残留的基因组DNA。反转录按SuperScript III First-Strand Synthesis system试剂盒说明进行。RNA操作过程均在超净台中进行,并使用无RNase的仪器设备操作。
1.2.4 qRT-PCR和半定量RT-PCR qRT-PCR按SYBR Green的反应体系在Light Cycler480上进行,每个实验3个重复,使用的引物对分别为GAPDH:5′-GGTATCGTGGAAGGACTCATGAC-3′,5′-ATGCCAGTGAGCTTCCCGTTCAG-3′;zkscan1:5′-ATTCGTCTCGGAAACCCC-3′,5′-TCCCGTGATTCAGCAGTC-3′。半定量RT-PCR按照常规PCR反应进行,一般为22或24个循环取样电泳,使用的引物对分别为GAPDH:5′-CGGATTTGGTCGTATTGGG-3′,5′-TCTTGAGGCTGTTGTCATACTTCT-3′;zkscan1:5′-TCATGGACCTGAGATGCTCG-3′与5′-TACCATCCTTCTCCTGTGCA-3′或5′-AGTCCCACTTCAAACATTCGTCT-3′与 5′-CTGTTGTCCTGATAAATCAAGC-3′;ZNF608:5′-CGGTTGAACAGCTTTTGGTT-3′,5′-TGGATGAAGATGGGGAAAAG-3′;CAMSAP1:5′-TCAGTGCCTCGAAAGAACTTCCGT-3′,5′-TGTGCTCCTGCTCATACTGGTCAA-3′;HIPK3:5′-TATGTTGGTGGATCCTGTTCGGCA-3′,5′-TGGTGGGTAGACCAAGACTTGTGA-3′;clec2d:5′-CACCCTTGGAAGTGGACAGA-3′,5′-TGCTCTGACGACTCTCTGTG-3′;G3PDH:5′-ACCACAGTCCATGCCATCAC-3′,5′-TCCACCACCCTGTTGCTGTA-3′。
1.2.5 RNase R敏感性分析 典型的反应为20 μL总体系中加入2 μL RNase R 反应缓冲液、10 μg总RNA以及0.5 μL的RNase R,37℃反应2 h。对照组不加入RNase R,其他反应条件不变。反应结束后,取等体积(一般为1 μL)反应体系立刻进行RNA反转录和RT-PCR反应。通过分别比较实验组和对照组中的GAPDH或靶基因的表达差异判断其RNase R敏感性。
1.2.6 细胞药物处理[10,16,17]将α-鹅膏蕈碱固体粉末溶于ddH2O中配置为1 mg/mL的母液,将ML-60218固定粉末溶于DMSO中配置为20 mmol/L母液;为了消除药物对转染效率的影响,待转染过程完成6 h后(此时转染基本结束),将细胞换到含不同浓度药物的DMEM培养基继续培养12 h。体积不足的部分加ddH2O或DMSO补齐。
1.2.7 RNase H处理 15 μg 细胞总RNA与1 μg寡核苷酸探针混合于16 μL体系中,70℃孵育1 min后立刻置于冰上1 min以上,然后再加入2 μL 10×RNase H缓冲液混匀后,37℃孵育10 min,最后加入2 μL RNase H(5 U/μL),37℃孵育30 min。
1.2.8 Northern Blot 通过甲醛变形琼脂糖凝胶电泳分离细胞总RNA后转印至尼龙膜,紫外交联后使用标记好的放射性同位素杂交,通过ULTRAhyb-Oligo杂交液42℃预杂交30 min后加入标记好的放射性探针42℃杂交过夜。寡核苷酸探针的放射性标记总体系为20 μL,含2 μL32γ-ATP(10 mCi/mL),1 μL寡核苷酸(10 μmol/L),1 μL T4 PNK(10 U/μL),37度标记1 h。使用50 mL洗膜液(2×SSC,0.5% SDS)42℃洗膜2-3次,最后将尼龙膜取出后用滤纸干燥,用保鲜膜包住放入含磷屏的暗夹中曝光12-24 h,通过Typhoon scanner扫描磷屏信号。根据参考文献[18]设计了合适的寡核苷酸探针,探针1:5′-GCGTTGGCGGAATATCTCTGGGTC-3′;探针 2:5′-TTTACTATTCCTCGTGACTG-3′;β-Actin:5′-AGCACTGTGTTGGCGTACAG-3′。
2 结果
2.1 外源DNA在哺乳动物细胞中不依赖于启动子的转录和转录本剪接的现象
为了研究circRNA的生成机制,使用人zkscan1基因位置的环状RNA作为研究对象。参与环化的外显子以及其上下游约1 kb内含子序列克隆至真核表达载体pcDNA3.1中(pcDNA3.1_zkscan1,图1-A)。将表达载体转入293T细胞后,通过荧光定量PCR,可以检测到丰富的反向剪接产物(图1-B)。而在实验偶然转染的原核克隆中间载体(pGEMT_zkscan1)中也能检测到同样丰富的产物(图1-B)。
为了排除可能存在的真核表达元件,将zkscan1序列区域以正向(pGEMT_zkscan1_F)或反向(pGEMT_zkscan1_R)插入pGEMT载体后转入细胞,通过半定量RT-PCR均能检测到“反向剪接产物”(图1-C)。为了进一步排除载体骨架序列的影响,只将覆盖zkscan1区域的PCR片段转染入细胞后的结果(图1-C)显示,丰富的转录产物依然能被检测到,说明载体序列不参与转录。
为了进一步验证这些产物是否具有环状构型,通过RNase R(仅能降解线性RNA而不能降解环状RNA的外切酶)处理转染不同质粒细胞的总RNA后进行半定量RT-PCR(图1-D)发现,转染了pcDNA3.1_zkscan1质粒细胞中的反向剪接产物几乎不受RNase R的影响,符合预期,而转染了pGEMT_zkscan1载体细胞中的反向剪接产物几乎完全被降解,说明后者表达的大部分产物可能没有环状构型。在本研究中,为了区别其与环状RNA的反向剪接方式,称这种特殊的剪接为“异常剪接”。
2.2 通过Northern blot分析不依赖于启动子的转录产物
为了进一步分析这些异常剪接产物,使用了两个探针做Northern blot,探针1识别外显子2,探针2设计在跨反向剪接位点上,前者能检测到含有外显子2的所有RNA分子,包括线性mRNA分子和环状RNA,而后者只能检测到由外显子反向剪接产生的环状RNA(图2-A)。Northern blot结果(图2-B)显示,空载体不表达任何RNA分子,正对照400-1 882表达3种RNA分子。实验组中只有转染了pcDNA3.1_zkscan1质粒的细胞才能检测到相应的RNA分子,而转染了pGEMT_zkscan1或zkscan1 PCR扩增产物的组均不能检测到明显的条带。推测这些特殊的RNA分子可能具有很长的分子结构,因此在琼脂糖凝胶中积在胶孔附近难以转膜。

图1 外源zkscan1基因片段能进行不依赖于启动子的异常转录
当用探针1和RNase H水解转染了zkscan1 PCR扩增产物后的细胞总RNA后,以探针2进行Northern blot杂交可以检测到明显的条带,而探针1检测不到信号,反之亦然(图2-C)。这些结果表明,这些异常的转录产物确实存在,而且极有可能具有串联重复的长分子结构。
2.3 异常转录本的剪接依赖于细胞内源的剪接信号
通过RT-PCR实验分析转染了pcDNA3.1_zkscan1、pGEMT_zkscan1及zkscan1 PCR扩增产物的细胞总RNA发现,它们存在相同的扩增图谱,共产生3种剪接产物(图4-A,WT)。而将下游内含子剪接位点的GU突变为CA后,均发现了同样的剪接条带的上移(图4-A,Mut)。Sanger测序显示,这些不同条带的产物均符合“GU-AG”法则,说明异常转录本的产生也是利用了同样的细胞内源剪接信号(图4-B)。
2.4 RNA聚合酶II参与异常转录
利用真核生物RNA聚合酶对α- 鹅膏蕈碱和ML-60218的敏感性,研究这种不依赖于启动子的特殊转录是由哪一种RNA聚合酶负责合成。真核生物RNA聚合酶II对α-鹅膏蕈碱高度敏感,是RNA聚合酶III的敏感性的100倍。ML-60218是RNA聚合酶III的特异性抑制剂,抑制能力IC50为27 μmol/L,对其他两种酶没有抑制作用。如图4-A显示,当α-鹅膏蕈碱的浓度为1 μg/mL时,只抑制RNA聚合酶II,几乎不抑制RNA聚合酶III,不依赖于启动子的转录(pGEMT_zkscan1_异常剪接)被很大程度抑制,而且这种抑制效应等同于RNA聚合酶II类型的启动子CMV起始的转录反应(pcDNA_zkscan1_反向剪接)。与α-鹅膏蕈碱的有效抑制作用不同,高浓度的ML-60218(> 2倍IC50)则对两种转录都没有明显的抑制作用(图4-B)。推测RNA聚合酶II极有可能负责这种不依赖于启动子的转录。
2.5 不依赖于启动子的DNA转录和剪接在哺乳动物细胞中是保守的
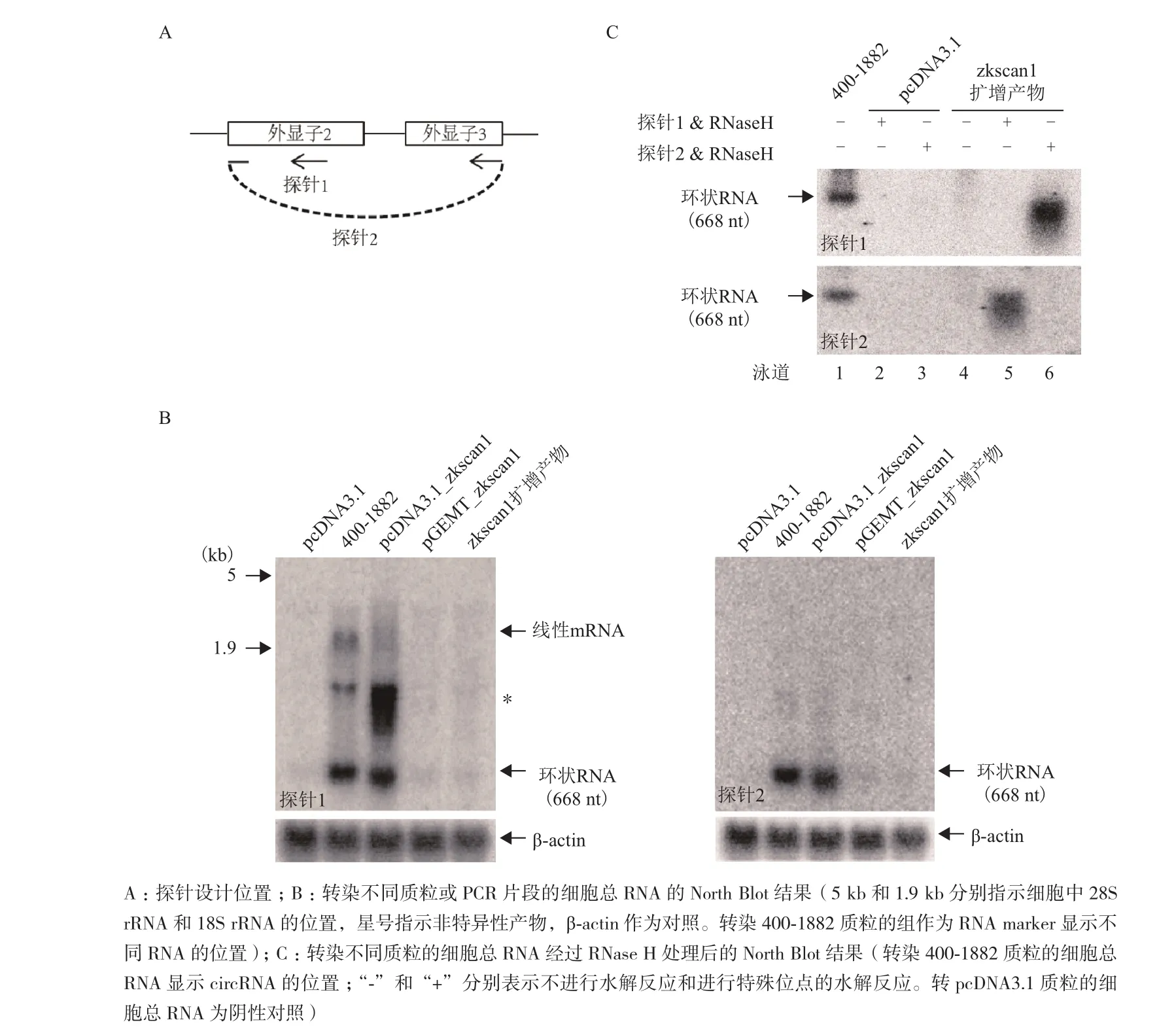
图2 Northern blotting鉴定不同的转录产物
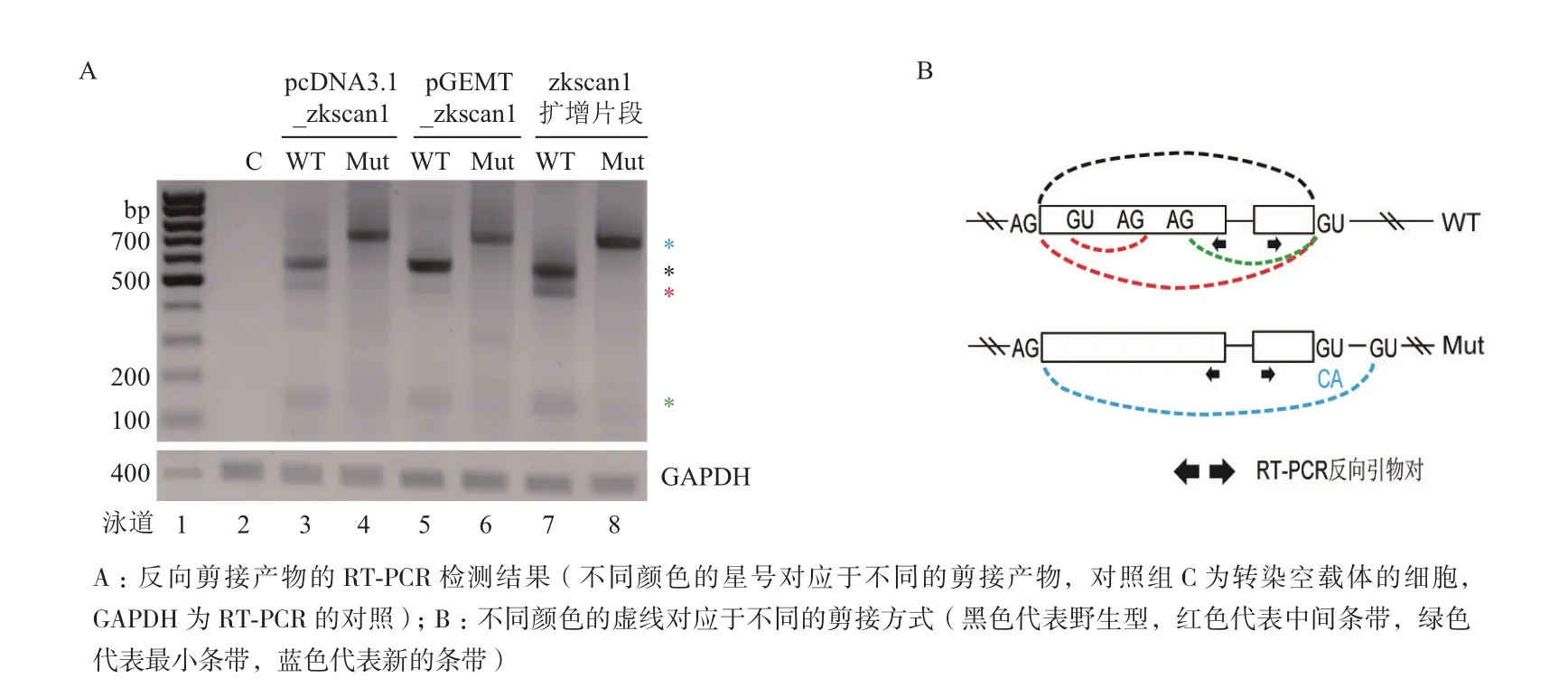
图3 转染不同质粒后对应的反向/异常剪接产物的剪接模式
为了验证这种不依赖于启动子的DNA转录和剪接的保守性,从人基因组中克隆了产生环状RNA的外显子DNA区段以及其上下游部分内含子序列(ZNF608,CAMSAP1,HIPK3)。分别在不同的PCR循环数时取5 μL RT-PCR产物进行电泳分析。和内源反向剪接产物需要在30-35个PCR循环数时出现扩增条带相比,转染外源DNA的异常剪接产物均能在18-22个PCR循环数时出现扩增条带(图5-A),说明它们均高效表达。

图 4 RNA聚合酶II负责不依赖于启动子的转录
当将pcDNA3.1_zkscan1和pGEMT_zkscan1载体导入小鼠NIH 3T3细胞后,半定量RT-PCR结果显示前者的反向剪接以及后者的异常剪接现象均能观察到。当将一段小鼠基因片段(clec2d)按同样的方法克隆转入人293T细胞后,也能观察到小鼠基因在人细胞中的异常剪接现象。这些结果表明哺乳动物细胞中不依赖于启动子的DNA转录和剪接是保守的。
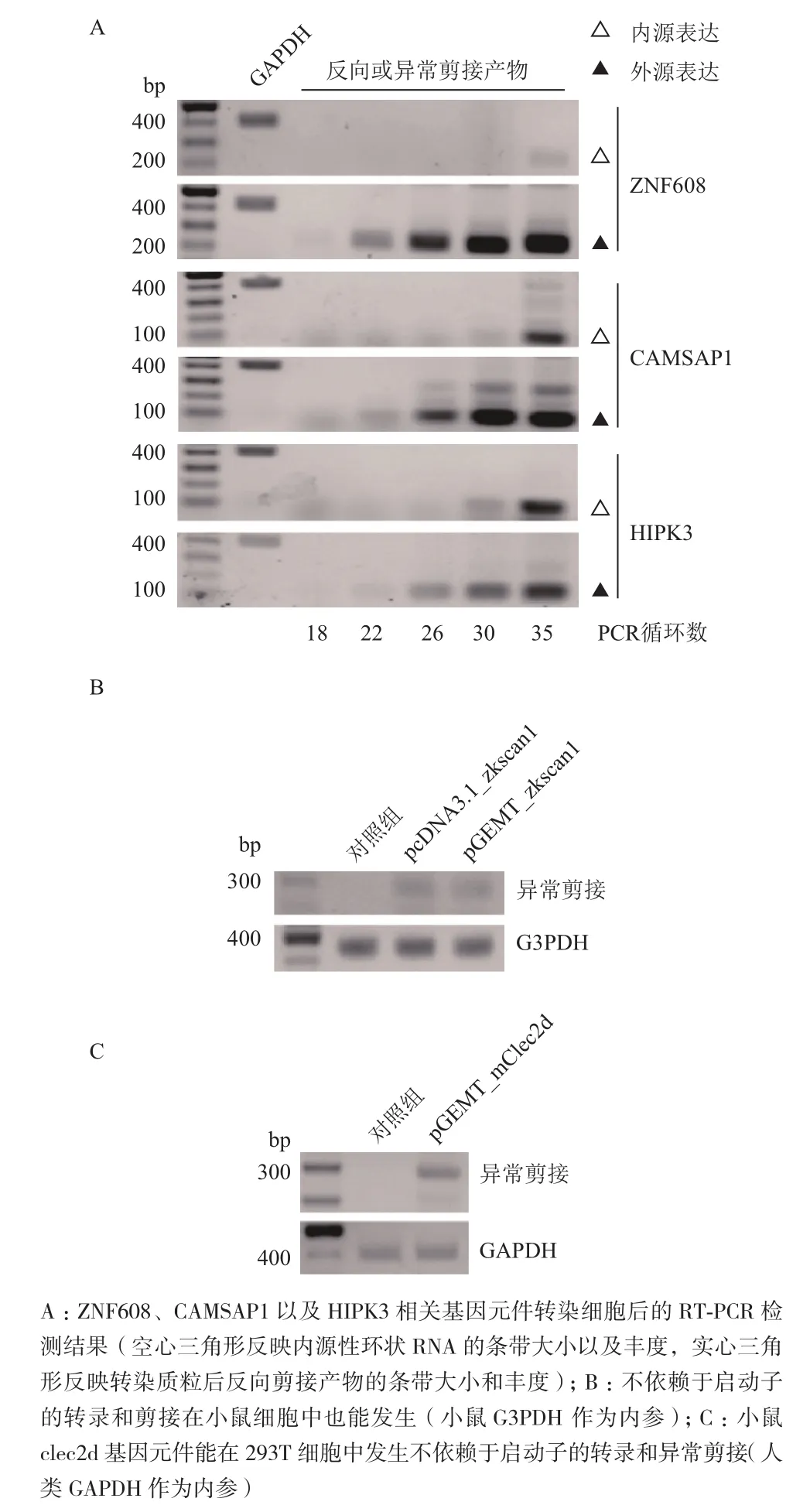
图5 不依赖启动子的转录与剪接没有序列选择性
3 讨论
本研究证实了含外显子的双链DNA分子导入哺乳动物细胞后能被RNA聚合酶识别和有效转录。由于转染的外源DNA分子整合到宿主基因组的概率极低(千分之一或更低),这些异常转录本很可能是来源于游离于染色体外的DNA分子的转录[5]。根据RNA聚合酶的抑制性实验结果,推测RNA聚合酶II很有可能参与这种异常的转录。一般情况下,RNA聚合酶II通过识别特殊的启动子区域进行下游DNA的转录。根据报道,细胞内源的基因组DNA大多以染色质形式存在,经过复杂的DNA修饰或结合多种组蛋白[19,20]。而导入细胞的双链DNA是提取自原核生物的质粒或通过PCR扩增的片段,均处于裸露状态,RNA聚合酶更容易结合。本研究的发现证明,细胞从外源获取的DNA分子除了通过水平基因转移对宿主细胞产生影响,还有可能在RNA水平对宿主细胞产生更直接的影响。
同时,本研究还对环状RNA的表达分析有重要发现,即通过反向引物对进行RT-PCR检测到的分子并不一定是以环状构型存在,还需要进行RNase R的进一步验证。
4 结论
本研究使用真核表达载体构建了环状RNA的表达体系,并发现了外源DNA在哺乳动物细胞中不依赖于启动子的转录和转录本的剪接现象;证明了这种现象在哺乳动物细胞中是保守的。该研究对环状RNA的表达分析具有重要的借鉴作用。
[1]Perucho M, Hanahan D, Wigler M. Genetic and physical linkage of exogenous sequences in transformed cells[J]. Cell, 1980, 22:309-317.
[2]Folger KR, Wong EA, Wahl G, et al. Patterns of integration of DNA microinjected into cultured mammalian cells:evidence for homologous recombination between injected plasmid DNA molecules[J]. Mol Cell Biol, 1982, 2:1372-1387.
[3] Wilson JH, Berget PB, Pipas JM. Somatic cells efficiently join unrelated DNA segments end-to-end[J]. Mol Cell Biol, 1982, 2:1258-1269.
[4]Bandyopadhyay PK, Watanabe S, Temin HM. Recombination of transfected DNAs in vertebrate cells in culture[J]. Proc Natl Acad Sci USA, 1984, 81:3476-480.
[5]Biamonti G, Della VG, Talarico D, et al. Fate of exogenous recombinant plasmids introduced into mouse and human cells[J]. Nucleic Acids Res, 1985, 13:5545-561.
[6]Murnane JP, Yezzi MJ, Young BR. Recombination events during integration of transfected DNA into normal human cells[J]. Nucleic Acids Res, 1990, 18:2733-2738.
[7]Chang XB, Wilson JH. Modification of DNA ends can decrease end joining relative to homologous recombination in mammalian cells[J]. Proc Natl Acad Sci USA, 1987, 84:4959-4963.
[8]Carroll D, Wright SH, Wolff RK, et al. Efficient homologous recombination of linear DNA substrates after injection into Xenopus laevis oocytes[J]. Mol Cell Biol, 1986, 6:2053-2061.
[9]Powell DJ, Galli C, Moor RM. The fate of DNA injected into mammalian oocytes and zygotes at different stages of the cell cycle[J]. J Reprod Fertil, 1992, 95:211-220.
[10] Seidl CI, Lama L, Ryan K. Circularized synthetic oligodeoxynucleotides serve as promoterless RNA polymerase III templates for small RNA generation in human cells[J]. Nucleic Acids Res, 2013, 41:2552-2564.
[11]Lama L, Seidl CI, Ryan K. New insights into the promoterless transcription of DNA coligo templates by RNA polymerase III[J]. Transcription, 2014, 5:e27913.
[12]Nejepinska J, Malik R, Moravec M, et al. Deep sequencing reveals complex spurious transcription from transiently transfected plasmids[J]. PLoS One, 2012, 7:e43283.
[13]Lasda E, Parker R. Circular RNAs:diversity of form and function[J]. RNA, 2014, 20:1829-1842.
[14]Jeck WR, Sorrentino JA, Wang K, et al. Circular RNAs are abundant, conserved, and associated with ALU repeats[J]. RNA, 2013, 19:141-157.
[15]Guo JU, Agarwal V, Guo H, et al. Expanded identification and characterization of mammalian circular RNAs[J]. Genome Biology, 2014, 15:409.
[16]Bensaude O. Inhibiting eukaryotic transcription:Which compound to choose? How to evaluate its activity?[J]. Transcription, 2011, 2:103-108.
[17]Wu L, Pan J, Thoroddsen V, et al. Novel small-molecule inhibitors of RNA polymerase III[J]. Eukaryotic Cell, 2003, 2:256-264.
[18]Liang D, Wilusz JE. Short intronic repeat sequences facilitate circular RNA production[J]. Genes & Development, 2014, 28:2233-2247.
[19]Heslop-Harrison JS, Schwarzacher T. Nucleosomes and centromeric DNA packaging[J]. Proceedings of the National Academy of Sciences of the United States of America, 2013, 110:19974-195.
[20]Wolff JA, Budker V. The mechanism of naked DNA uptake and expression[J]. Advances in Genetics, 2005, 54:3-20.
(责任编辑 李楠)
Promoter-independent Transcription of Exogenous DNA and Subsequent RNA Splicing in Mammalian Cells
XU Wen1YANG Hua-yu2ZHENG Yong-chang2
(1. School of Life Sciences,Tsinghua University,Beijing 100084;2. Department of Liver Surgery,Peking Union Medical College Hospital,Chinese Academy of Medical Sciences and Peking Union Medical College,Beijing 100730)
Introduction of exogenous double stranded DNA into mammalian cells will undergo restructuring,rearrangement and other complex molecular and biochemical reaction. Few DNA molecules will randomly integrate into the host genome,namely the occurrence of horizontal gene transfer. However,it is unclear whether these exogenous DNA molecules can be recognized by cell machine and triggers the transcription in the circumstance of containing no special promoter. During the construction of circle RNA(circRNA)expression system,we discovered that promoter-independent transcription of exogenous DNA occurred,also RNA splicing was in accord with “GU-AG” in transcripts,and subsequently abnormal spliced molecules similar to the circRNA formed. Further studies showed that the promoter-independent transcription and splicing of exogenous DNA in mammalian cells were conservative. Conclusively,this work revealed a new destiny approach of exogenous DNA in the mammalian cells,which is of significance and applicability in studying the effects of exogenous DNA on host cells.
promoter-independent transcription;RNA splicing;circRNA;DNA transfection;alternative splicing
10.13560/j.cnki.biotech.bull.1985.2017-0183
2017-03-10
国家高技术研究发展计划(2015AA020303)
徐文,男,硕士研究生,研究方向:分子生物学;E-mail:xuwenhbnu@163.com
郑永昌,男,副主任医师,研究方向:肝胆肿瘤分子生物学;E-mail:zyc_pumc@163.com
