清热解毒胶囊质量研究
2017-07-31刘志荣李安平杨平荣胡旭东
刘志荣 李安平 杨 锡 杨平荣 胡旭东
(甘肃省药品检验研究院,兰州 730070)
清热解毒胶囊质量研究
刘志荣 李安平 杨 锡 杨平荣 胡旭东
(甘肃省药品检验研究院,兰州 730070)
目的:完善提高清热解毒胶囊的质量标准。方法:采用薄层色谱法对处方中金银花、栀子、连翘、麦冬、龙胆、黄芩进行定性鉴别;同时测定处方中绿原酸和黄芩苷含量。采用CAPCELL PAK C18(5 μm,4. 6 mm ×250 mm)为色谱柱,以乙腈-0.1%磷酸水溶液为流动相,梯度洗脱,流速1 mL·min-1,检测波长326 nm,柱温30 ℃。结果:薄层色谱鉴别专属性强,重复性好。样品中的绿原酸和黄芩苷峰按照拟定的试验方法测定分离度良好,分别在0.0479μg~0.957 μg(r=0.9999)、0.153μg~3.064 μg(r=0.9999)范围内与峰面积呈现良好的线性关系;加样回收率分别为92.7%(RSD=1.9)和98.9%(RSD=2.0)。经HPLC测定,10批样品中2个成分的含量分别为绿原酸3.74~4.51 mg·g-1,黄芩苷18.94~21.76 mg·g-1。结论:该法简便、专属、准确,可用于清热解毒胶囊的质量控制。
清热解毒胶囊 TLC HPLC 绿原酸 黄芩苷
清热解毒胶囊处方由石膏、金银花、玄参、地黄、连翘、栀子、甜地丁、黄芩等12味中药材组成,主要功能为清热解毒,养阴生津,泻火,可用于风热型感冒、流行性腮腺炎及轻、中型乙型脑炎等症[1,2]。现行标准中仅有理化鉴别项,在质量控制中尚显不足,如鉴别部分仅以薄层色谱鉴定了栀子、金银花、连翘3味药味,其余药味都没有设置相应的检测项目[3]。而含量测定项下仅采用高效液相色谱法测定黄芩中黄芩苷的含量[3],该标准已经难以适应新形势下从严、综合控制药品质量的要求。文献报道[4-7]清热解毒胶囊中黄芩苷、连翘苷等的含量测定较多,但关于薄层鉴别方面的文献报道较少。为更好的控制药品质量,本实验在原有标准的基础上,增加了麦冬、龙胆的薄层色谱定性鉴别;增加了用HPLC法同时测定处方中绿原酸和黄芩苷的含量,并进行相关的方法学验证。
1 仪器与试药
岛津LC-20A型高效液相色谱仪(配备SPD-20A检测器和LCsolution色谱工作站);资生堂CAPCELL PAK C18(5 μm,4. 6 mm ×250 mm)色谱柱;ME-204 电子天平,德国Sartorius R200D 型分析天平(十万分之一);KH-500DE超声波清洗器(功率250 W,频率40 kHz,上海超声波仪器厂)。绿原酸(批号:110753-201415)、黄芩苷(批号:110715-201318)、麦冬(批号:121013-201310)、龙胆(批号:121530-200501)、黄芩(批号:120955-201309)、连翘(批号:120908-201216)、栀子(批号:120986-201309)、金银花(批号:121060-201107)均购自中国药品生物制品鉴定所;清热解毒胶囊样品由陕西步长制药有限公司提供,图中记为胶囊(批号);乙腈(色谱纯),其他试剂均为分析纯,实验用水为超纯水。
2 实验方法与结果
2.1 薄层色谱定性鉴别
2.1.1 金银花的定性鉴别
取清热解毒胶囊内容物2.0 g,加甲醇5 mL,超声处理30 min,过滤,滤液作为供试品溶液。另取金银花对照药材0.2 g,同法制成对照药材溶液。吸取上述两种溶液各5 μL,分别点于同一硅胶G薄层板上,以乙酸丁酯-甲醇-水(7∶2.5∶2.5)的上层液为展开剂,展开,取出,晾干,置紫外光灯(365 nm)下检视。供品试品色谱图中,在与对照药材色谱图相应的位置上,显相同颜色的荧光斑点,阴性对照品在此处无干扰,见图1。

图1 金银花样品及不同批号胶囊的薄层色谱图1.金银花对照药材;2. 缺金银花阴性样;3. 胶囊(160301);4. 胶囊(160210);5. 胶囊(150503)
2.1.2 栀子的定性鉴别
取清热解毒胶囊内容物2.0 g,加50%甲醇10 mL,超声处理40 min,滤过,滤液作为供试品溶液。另取栀子对照药材0.2 g,同法制成对照药材溶液。吸取上述两种溶液各5 μL,分别点于同一硅胶G板上,以乙酸乙酯-丙酮-甲酸-水(5∶5∶1∶1)为展开剂,展开,取出,晾干,喷以10%硫酸乙醇溶液,105 ℃加热至斑点显色清晰。供品试品色谱图中,在与对照药材色谱图相应的位置上,显相同颜色的斑点,阴性对照品在此处无干扰,见图2。

图2 栀子样品及不同批号胶囊的薄层色谱图1.栀子对照药材;2. 缺栀子阴性样;3. 胶囊(160301);4. 胶囊(160210);5. 胶囊(150503)
2.1.3 连翘的定性鉴别
取清热解毒胶囊内容物2.0 g,加甲醇10 mL,超声处理20 min,滤过,滤液蒸干,残渣加2 mL甲醇溶解作为供试品溶液。另取连翘对照药材0.2 g,同法制成对照药材溶液。吸取上述两种溶液各5~10 μL,分别点于同一硅胶G薄层板上,以三氯甲烷-甲醇(8∶1)为展开剂,展开,取出,晾干,喷以10%硫酸乙醇溶液,105 ℃加热至斑点显色清晰。供品试品色谱图中,在与对照药材色谱图相应的位置上,显相同颜色的斑点,阴性对照品在此处无干扰,见图3。

图3 连翘样品及不同批号胶囊的薄层色谱图1.连翘对照药材;2. 缺连翘阴性样;3. 胶囊(160301);4. 胶囊(160210);5. 胶囊(150503)
2.1.4 麦冬的定性鉴别
取清热解毒胶囊内容物2.0 g,加三氯甲烷-甲醇(7∶3)混合溶液10 mL,浸泡3 h后超声处理30 min,滤过,滤液蒸干,残渣加2 mL三氯甲烷溶解,作为供试品溶液。另取麦冬对照药材1 g,同法制成对照药材溶液。吸取上述两种溶液各2~5 μL,分别点于同一硅胶G薄层板上,以甲苯-甲醇-冰醋酸(80∶5∶0.1)为展开剂,展开,取出,晾干,置紫外光灯(254 nm)下检视。供品试品色谱图中,在与对照药材色谱图相应的位置上,显相同颜色的荧光斑点,阴性对照品在此处无干扰,见图4。

图4 麦冬样品及不同批号胶囊的薄层色谱图1.麦冬对照药材;2. 缺麦冬阴性样;3. 胶囊(160301);4. 胶囊(160210);5. 胶囊(150503)
2.1.5 龙胆的定性鉴别
取清热解毒胶囊内容物2.0 g,加甲醇10 mL,超声处理20 min,滤过,滤液蒸干,残渣加2 mL甲醇溶解,作为供试品溶液。另取龙胆对照药材0.5g,同法制成对照药材溶液。吸取上述两种溶液各5 μL,分别点于同一硅胶GF254薄层板上,以乙酸乙酯-甲醇-水(10∶2∶1)为展开剂,展开,取出,晾干,置紫外光灯(254 nm)下检视。供品试品色谱图中,在与对照药材色谱图相应的位置上,显相同颜色的荧光斑点,阴性对照品在此处无干扰,见图5。

图5 龙胆样品及不同批号胶囊的薄层色谱图1.龙胆对照药材;2.缺龙胆阴性样;3. 胶囊(160301);4. 胶囊(160210);5. 胶囊(150503)
2.1.6 黄芩的定性鉴别
取清热解毒胶囊内容物2.0 g,加50%甲醇10 mL,超声处理30 min,滤过,滤液蒸干,加2 mL甲醇溶解作为供试品溶液。另取黄芩对照药材0.2 g,同法制成对照药材溶液。吸取上述两种溶液各5 μL,分别点于同一硅胶G薄层板上,以乙酸乙酯-丁酮-甲醇-水(15∶3∶1∶1)为展开剂,展开,取出,晾干,喷以1%三氯化铁乙醇溶液。供品试品色谱图中,在与对照药材色谱图相应的位置上,显相同颜色的斑点,阴性对照品在此处无干扰,见图6。
2.2 HPLC定量测定
2.2.1 色谱条件

图6 黄芩样品及不同批号胶囊的薄层色谱图1.黄芩对照药材;2. 缺黄芩阴性样;3. 胶囊(160301);4. 胶囊(160210);5. 胶囊(150503)
色谱柱为资生堂CAPCELL PAK C18(5 μm,4.6 mm ×250 mm),以乙腈-0.1%磷酸水溶液为流动相梯度洗脱(见表1),检测波长为326 nm,柱温30 ℃,流速:1.0 mL·min-1,进样量:10 μL。

表1 梯度洗脱程序
2.2.2 供试品溶液的制备
取供试品约0.3 g,精密称定,置100 mL三角瓶中,精密加入甲醇50 mL,称量,超声提取20 min,放冷,再称量,用甲醇补足减失的量,摇匀,滤过,取续滤液即得。
2.2.3 混合对照品溶液的制备
取绿原酸对照品和黄芩苷对照品适量,加甲醇制成每1 mL含绿原酸50 μg、黄芩苷150 μg,即得。
2.2.4 干扰试验
按处方比例及制备工艺,配制不含金银花和黄芩的阴性样品,并按供试品溶液制备方法制成阴性样品溶液。在上述色谱条件下,取供试品溶液、对照品溶液及阴性对照溶液各20 μL,注入液相色谱仪,结果表明其他成分对绿原酸和黄芩苷的测定无干扰。结果见图7。
2.2.5 线性范围
精密吸取对照品混合溶液1、3、5、10、15、20、25 μL,注入液相色谱仪,测定峰面积积分值,以峰面积积分值为横坐标(X),对照品溶液的进样量(μg)为纵坐标(Y)绘制标准曲线,计算回归方程,结果见表2。结果表明,绿原酸和黄芩苷分别在0. 0479~0.957 μg、0.153~3.064 μg范围内具有良好的线性关系。

图7 供试品对照品高效液相色谱图a.对照;b.供试品;c.金银花阴性对照;d.黄芩苷阴性对照

成分回归方程r线性范围(μg)绿原酸Y=2.981×10-7-0.0010.99990.0479~0.957黄芩苷Y=4.750×10-7-0.0020.99990.153~3.064
2.2.6 精密度实验
精密吸取绿原酸和黄芩苷混合对照品溶液10 μL,注入液相色谱仪,连续进样6次,测定峰面积积分值,计算相对标准偏差,结果见表3。绿原酸和黄芩苷峰面积的RSD均为0.4%,小于3%,符合中国药典的相关要求。

表3 进样精密度测定结果(A:峰面积)
2.2.7 稳定性试验
精密吸取同一供试品溶液10 μL,注入液相色谱仪,测定待测成分峰面积积分值,结果供试品中绿原酸和黄芩苷峰的积分面积在1950 min内RSD分别为0.5%、0.4%。稳定性符合中国药典的有关规定。结果见表4。

表4 稳定性试验结果(A:峰面积)
2.2.8 重复性实验
取同一批号样品按上文拟订的制备和测定方法平行测定6次,绿原酸及黄芩苷重复性实验结果较好,符合中国药典的有关规定,结果见表5。

表5 样品测定结果的重复性(批号:150503)
2.2.9 加样回收率试验
取已知绿原酸、黄芩苷含量的供试品(批号:150503)9份,每份约0.15 g,精密称定,置锥形瓶中,分别精密加入一定量的混合对照品溶液(绿原酸0.1542 mg·mL-1、黄芩苷0.7621 mg·mL-1),按供试品溶液制备方法制备,测定,绿原酸、黄芩苷的回收率结果复合中国药典相关规定,表明该含量测定方法可行(见表6、表7)。

表6 绿原酸加样回收率试验结果

表7 黄芩苷加样回收率试验结果
2.2.10 供试品含量测定
按上述方法测定10批样品,结果见表8。
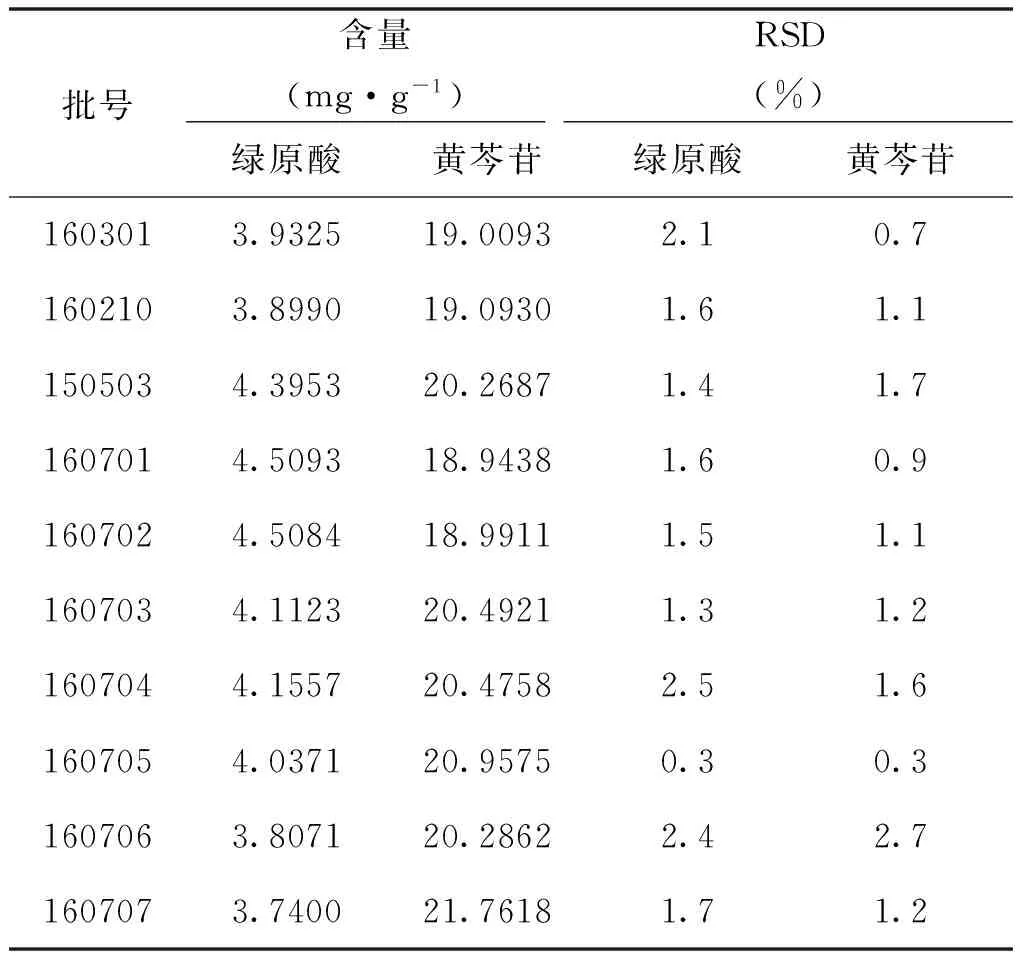
表8 样品测定结果
3 讨论
3.1 供试品提取条件的优化
分别选择超声波提取、回流提取两种提取方法进行实验考察,结果表明,超声波提取效率最高,且简便快捷,故实验选用超声提取法制备供试品溶液。对提取溶剂(甲醇、50%甲醇)进行优选,发现甲醇提取供试品中各检测组分总含量优于50%甲醇提取,且杂质明显减少,因此选择甲醇作为该供试品的提取溶剂。供试品量相同情况下对提取溶剂用量(25 mL、50 mL和100 mL)进行优化,发现50 mL和100 mL用量下各检测组分总含量基本相同,均优于25 mL提取液,因此选择50 mL作为该供试品的提取溶剂量。
3.2 波长的选择
由于清热解毒颗粒组成处方较大,化学成分复杂而呈多样性,本研究拟同时测定绿原酸和黄芩苷两种成分,使得一次检测就可以控制处方中金银花及黄芩两味药的含量。由于上述成分的紫外吸收情况差距较大,通过比较两种成分在200~400 nm 的紫外光谱发现,绿原酸和黄芩苷的最大吸收波长分别为326 nm和276 nm,特征吸收的差异较大,综合考虑并比较不同波长下其色谱峰的分离情况和强度,本试验选择326 nm 作为检测波长。结果显示,在此条件下两 种成分的峰面积响应及线性关系良好,符合含量测定的要求。同时,采用单波长同时测定制剂中绿原酸和黄芩苷的含量,较多波长方法测定更为简便,对仪器的要求较低,有利于测定工作的开展和实现。
3.3 流动相的选择
对不同流动相的组成比例进行考察,采用不同比例的甲醇-水系统、甲醇-醋酸水系统、甲醇-磷酸水系统、乙腈-水系统、乙腈-醋酸水系统和乙腈-磷酸水系统,发现在流动相中加入一定比例的酸有助于提高待测成分与干扰峰的分离度、改善峰形,经过进一步考察,确定采用由乙腈-0.1%磷酸水溶液组成的流动相系统进行梯度洗脱,可获得理想的色谱图。
4 总结
本实验改进并增加了清热解毒胶囊中药味的薄层定性鉴别方法,并且建立了高效液相色谱法同时测定清热解毒胶囊中绿原酸和黄芩苷的方法。绿原酸和黄芩苷的含量测定方法前处理条件简便、快速,所建立的梯度洗脱方法灵敏、准确,测定结果稳定可靠,重复性好,实用性强。本实验实现了从多成分、系统化角度控制清热解毒胶囊的质量。
[1]宋恩峰, 汪六林, 宋金春. 清热解毒颗粒防治感冒疗效观察[J]. 陕西中医, 2013 (12):1584-1586.
[2]隋继成, 孙志云. 清热解毒颗粒抗炎作用的实验研究[J]. 河南中医, 2008, 28(11):42-43.
[3]国家药典委员会. 国家药品标准新药转正标准. 中药第四十一册[S]. 2004: 91.
[4]李翔, 刘皈阳, 马建丽, 等. HPLC 法同时测定清热解毒软胶囊中绿原酸栀子苷和黄芩苷的含量[J]. 解放军药学学报, 2013 (3): 239-241.
[5]费毅琴, 聂晶. HPLC 法同时测定清热解毒软胶囊中黄芩苷和栀子苷的含量[J]. 中国药事, 2012, 26(5): 490-493.
[6]郭琪, 闫艳, 王海波, 等. HPLC 波长切换法同时测定清热解毒颗粒中 8 个成分的含量[J]. 药物分析杂志, 2015, 35(9): 1601-1605.
[7]林远凤, 黄燕萍. HPLC 同时测定清热解毒胶囊中 6 种成分的含量[J]. 中国实验方剂学杂志, 2015, 21(17): 67-70.
Quality study on Qingre Jiedu capsule.
Liu Zhirong, Li Anping, Yang Xi, Yang Pingrong, Hu Xudong
(GansuProvincialInstituteofDrugControl,Lanzhou730070,China)
Honeysuckle, Fructus Gardeniae, Fructus Forsythiae, Radix Ophiopogonis, Radix Gentianae and Scutellaria Baicalensis were indentified by TLC. The content of chlorogenic acid and baicalin were determined by HPLC. The chromatographic separation was carried out on a CAPCELL PAK C18column (5 μm,4.6 mm×250 mm)with a mobile phase consisting of acetonitrile-0.1% phosphoric acid solution in a gradient mode at a flow rate of 1.0 mL·min-1. The UV detection wavelength was set at 326 nm while the column temperature was at 30℃.There was a good linear relationship within the range of 0.0479-0.957μg (r = 0. 9999) and 0.153-3.064 μg (r = 0. 9999) for chlorogenic acid and baicalin, respectively. The average recoveries of the two components were 92.7% (RSD = 1.9%) and 98.9% (RSD = 2.0%), respectively. The method is simple, exclusive, accurate and can be used for determination and quality control of qingrejiedu capsules.
Qingre Jiedu capsule; TLC; HPLC; chlorogenic acid; baicalin
国家标准提高项目(53)
10.3969/j.issn.1001-232x.2017.03.017
2016-12-08
