绿豆分子遗传图谱构建及若干农艺性状的QTL定位分析
2017-07-25王建花张耀文程须珍王丽侠
王建花张耀文程须珍王丽侠
1山西大学, 山西太原 030006;2中国农业科学院作物科学研究所, 北京 100081;3山西省农业科学院作物科学研究所, 山西太原030031
绿豆分子遗传图谱构建及若干农艺性状的QTL定位分析
王建花1,2,3张耀文3程须珍2,*王丽侠2,*
1山西大学, 山西太原 030006;2中国农业科学院作物科学研究所, 北京 100081;3山西省农业科学院作物科学研究所, 山西太原030031
利用花叶1号×紫茎1号杂交后代衍生的208个F2家系组建群体, 构建含有95个SSR标记位点的遗传连锁图谱, 该图谱包含11个连锁群, 全长1457.47 cM, 标记平均间距为15.34 cM。利用复合区间作图法, 对株高、幼茎色、主茎色、生长习性、结荚习性、复叶叶形和成熟叶色等农艺性状进行QTL分析, 分别检测到与株高、幼茎色、主茎色、复叶叶形有关的QTL各1个, 贡献率在8.49%~66.64%之间; 与结荚习性有关的QTL3个, 贡献率在60.32%~80.36%之间; 与成熟叶色有关QTL 4个, 贡献率在69.06%~87.35%之间; 与生长习性有关的QTL数量最多, 共26个, 贡献率在58.32%~99.51%之间。上述QTL主要分布在LG1、LG2、LG4、LG8和LG10连锁群, 其中LG1最少, 仅检测到生长习性的1个QTL, LG4最多, 包含了幼茎色、主茎色、结荚习性、生长习性、复叶叶形、成熟期叶色6个农艺性状的15个QTL; 这些QTL既可以应用于绿豆育种的分子标记辅助选择, 也对深入研究这些性状的遗传奠定了基础。
绿豆; 连锁遗传图谱; SSR标记; 复合区间作图法; QTL; 贡献率
绿豆[Vigna radiata (L.) Wilczek](2n=2x=22)是豆科(Leguminosae)蝶 形 花 亚 科 (Papilionaceae)菜 豆 族(Phaseoleae)豇豆属(Vigna)的一个种, 是一种快速生长的热季豆类作物, 主要在亚洲发展中国家种植, 是世界上重要的豆类作物。绿豆的基因组较小, Kang等[1]使用MAKER pipeline[2]预测并通过测序得到绿豆的基因组为421 Mbp (占全基因组得到的80%), 与豆科模式植物百脉根和蒺藜苜蓿基因组(均约为 470 Mbp)大小相近[3], 并绘制了豇豆属的第一个基因组序列草图。
图谱构建与基因定位是绿豆基因组研究的重要环节,是基因克隆乃至基因组结构与功能研究的基础。目前, 绿豆的基因定位研究还处于起步阶段, 已定位的性状主要有抗白粉病和抗豆象, Humphry等[4]、Reddy[5]、Parinya等[6]、Chaitieng等[7]、Young等[8]都有相关报道, 其中Reddy[5]以3个抗白粉病绿豆株系(V4718、V4758、V4785)为研究材料, 证明抗白粉病基因是由单显性基因控制的,主要有 Pm1、Pm2等。梅丽等[9]、赵丹等[10]、钟敏[11]和吴传书等[12]利用Berken/ACC41为材料, 将抗豆象基因定位在第9连锁群上一个2.4 cM的区间内, 离其两侧的标记C220和Vr2-627的距离分别为0.7 cM和1.7 cM。此外,梅丽等[9]以Berken/ACC41为材料, 利用基于混合线性模型[13]的QTLNetwork 2.0[14]软件的复合区间作图法进行QTL分析,并检测出播种到出苗时间、播种到开花时间、生育期等9个农艺性状相关的33个加性效应位点(赵丹等[10]和钟敏[11])。
绿豆中除了产量相关性状外, 还有好多表型性状可用于品种纯度鉴定、特性鉴别等研究中, 如花青甙显色、叶形等等。本研究利用具有特异叶形的花叶1号和具有明显花青甙显色的紫花1号的后代群体, 对某些表型性状进行 QTL定位, 旨在尽可能发掘有利用价值的等位基因,将分子标记辅助选择用于育种实践, 以培育优质高产和多抗新品种; 另一方面可从众多绿豆种质资源中选择出有价值的材料。
1 材料与方法
1.1 分离群体材料
2015年选用紫茎1号为父本、花叶1号为母本, 配制杂交组合。以幼茎色鉴别真假杂种后, 将杂种F2种植在北京, 小区行长1 m, 株距10 cm, 田间对亲本及208个F2群体的株高(PH)、幼茎色(YSC)、主茎色(MSC)、生长习性(GH)、结荚习性(PHA)、成熟叶色(MLC)(注: 收荚时叶片的颜色)和复叶叶形(TLS)性状, 严格按照《绿豆种质资源描述规范和数据标准》[15]的分级标准统计分析(其中复叶叶形人为划分为花、半圆、圆3个等级)并转换为“1”、“2”、“3”。
1.2 SSR引物
SSR引物均基于 SSR富集文库, 利用磁珠富集法开发[15], 其中单核苷酸单元重复序列引物(Vr1) 2007对, 二核苷酸单元重复序列引物(Vr2) 1696对, 三核苷酸单元重复序列引物(Vr3) 1485对, 共5188对。均由北京赛百盛生物技术有限公司合成。
1.3 总DNA提取和PCR扩增
采集亲本以及 F2群体每个单株的幼嫩叶片, 分别放入液氮罐中备用。用 Retsch MM400混合型碾磨仪研磨,改良的CTAB法[16]提取基因组DNA, 紫外分光光度计法检测DNA的质量。
PCR扩增反应总体积10 μL, 含30 ng基因组DNA, 1×Taq缓冲液 (20 mmol L–1Tris-HCl, pH 8.8; 10 mmol L–1KCl; 10 mmol L–1(NH4)2SO4; 1.5 mmol L–1MgCl2; 0.1% Triton X-100), 1 mmol L–1dNTPs, 上下游引物各 0.25 μmol L–1和1 U Taq DNA聚合酶。PCR程序为95℃预变性5 min, 95℃变性30 s, 55℃退火45 s, 72℃延伸45 s, 进行35个循环, 最后72℃延伸5 min, 4℃保存。整个反应在东胜EDC-810 PCR扩增仪上进行。
用 8%的聚丙烯酰胺非变性凝胶电泳分离扩增产物,在200 V恒压下电泳, 根据SNP分子量的大小及差异带型的可辨程度调整电泳时间, 一般在1.0~1.5 h左右。将凝胶用蒸馏水洗涤2次后, 用0.2%的AgNO3溶液染色10 min,蒸馏水洗涤2次后用1.5% NaOH加0.5%甲醛溶液显影至条带清晰, 蒸馏水洗涤2次后读带。
1.4 连锁图谱的构建
根据电泳结果, 用 A、B、H、“-”表示标记的母本、父本、杂合、缺失。根据已知分子量的Marker (600 bp DNA marker)计算各多态性条带的分子量。将 SSR多态性带按亲本来源归类, 分别转化为“0”、“2”、“1”、“–1”。根据各位点在作图群体中的分离比, 采用JoinMap 4.0软件构建F2群体的遗传连锁图谱。
1.5 QTL定位分析
利用QTL IciMapping V4.0软件进行QTL的定位。采用区间作图法对基因型和性状进行分析, 将 Permutation次数设置为1000次, LOD≥3.68作为QTL的入选临界值。检测性状与作图标记之间的关系及 QTL位点在遗传连锁图谱上的位置, 估算出与农艺性状相关基因位点的贡献率等参数。最后利用JoinMap 4.0整合的MapChart输出图谱。
2 结果与分析
2.1 SSR标记的多态性分析
共筛选5188对SSR引物, 4281对引物有稳定的扩增产物, 亲本间有多态且扩增稳定的的引物99对。其中Vr1引物有效扩增1651对, 有效扩增率82.26%, 多态性引物64对; Vr2引物有效扩增 1309对, 有效扩增率 77.18%,多态性引物55对; Vr3引物有效扩增1321对, 有效扩增率88.96%, 多态性引物32对, 表明二核苷酸单元重复序列引物多态性比率(4.20%)最高, 单核苷酸单元重复序列引物的多态性比率(3.88%)次之, 三核苷酸单元重复序列引物多态性比率(2.42%)最低。另外还从豇豆SSR引物和绿豆公布序列引物中分别得到亲本多态性引物 1对和3对。
2.2 连锁图谱的构建
利用筛选的 99对引物分析 208个 F2群体后, 应用JoinMap 4.0软件进行连锁分析, 构建绿豆遗传连锁图谱。该图谱包含了 95个 SSR标记(其中Vr3-985、Vr2-657、Vr1-574和Vr2-1430未连锁上), 分属11个连锁群(LG), 总覆盖基因组长度 1457.47 cM, 各连锁群长度在 5.52~613.74 cM之间, 其中LG6最短, LG4最长。不同连锁群上包含的标记位点在 2~28个之间, 标记之间平均距离在1.38~21.92 cM之间。
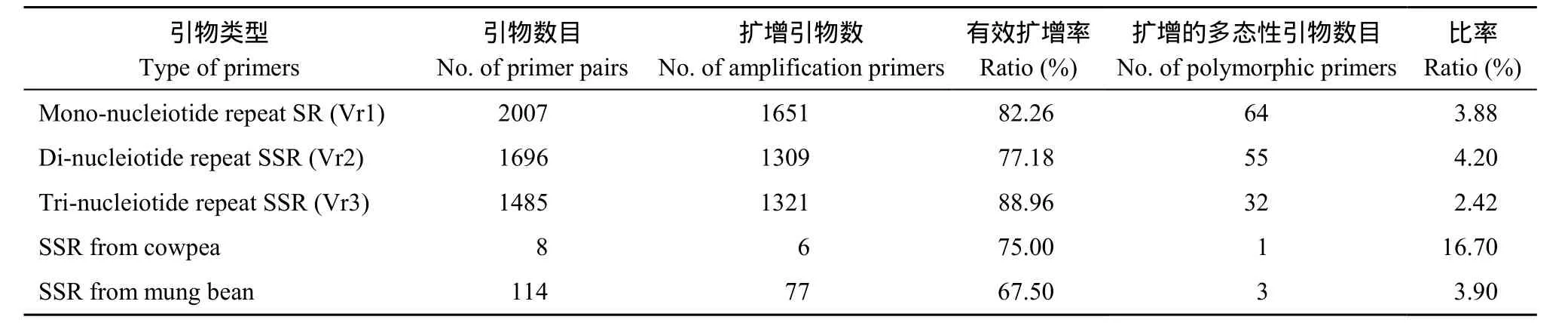
表1 不同引物在F2群体亲本中的扩增Table 1 Amplification of different markers in parents of F2

表2 遗传图谱分析Table 2 Analysis of genetic map
根据已知绿豆基因组引物标记分布情况, 比对本文连锁图谱可知, 第一连锁群(LG1)有11个标记, 其中只有Vr2-401位于绿豆基因组的Chr.6; LG2有19个标记, 其中70%的标记对应于绿豆基因组的Chr.9; LG3有7个标记,其中43%的标记对应于绿豆基因组的Chr.1; LG4有28个标记, 7个标记对应绿豆基因组的Chr.4, 8个对应绿豆基因组的Chr.5, 7个标记对应绿豆基因组的Chr.8; LG5有3个标记, 全都对应绿豆基因组的Chr.1; LG6有4个标记, 2个对应绿豆基因组的Chr.1, 另外2个对应Chr.7; LG7有2个标记, 对应绿豆基因组Chr.8; LG8有4个标记, 全都对应绿豆基因组Chr.7; LG9有5个标记, 其中3个对应绿豆基因组Chr.11; LG10有7个标记, 与绿豆基因组无明显的对应关系; LG11有5个标记, 全部对应绿豆基因组Chr.7。
2.3 绿豆重要农艺性状在F2群体中的表现
分析表明, 数量性状株高在F2群体中存在差异。各单株的株高差异明显, 标准差为 5.92, 变异幅度大, 且株高在F2群体中均呈正态分布(图1)。其余6个均为质量性状,且用 SAS统计软件对 F2群体 208个单株进行统计分析,株高(PH)、幼茎色(YSC)、主茎色(MSC)、生长习性(GH)、结荚习性(PHA)、复叶叶形(TLS)成熟叶色(MLC)的卡方检验结果(P=0.05)均不符合孟德尔遗传分离比(图1)。故可认为7个农艺性状均为多基因控制的遗传性状。
2.4 绿豆重要农艺性状的QTL定位
应用复合区间作图法, LOD等于3.68作为阈值。检测到7个性状的38个QTL (表3)。检测到1个株高QTL, 位于连锁群 8的 Vr3-932–Vr2-1233 (11.79 cM)区间, 与Vr2-1233的遗传距离为11.79 cM, 遗传贡献率为8.49%。
检测到1个幼茎色QTL, 位于连锁群4的Vr2-1042–Vr1-1057 (63.74 cM)区间, 与 Vr2-1042的遗传距离为28 cM, 遗传贡献率为66.64%。
检测到1个主茎色QTL, 位于连锁群4的Vr04-12148918–Vr2-1042 (45.49 cM)区间, 与Vr2-1042的遗传距离为19 cM, 遗传贡献率为38.68%。
检测到 3个结荚习性 QTL, 一个位于连锁群 2的Vr3-837–Vr2-440 (25.54 cM)区间, 与Vr2-440的遗传距离为9.7 cM, 遗传贡献率为60.32%; 另外2个都位于连锁群4的Vr1-545–Vr3-1177 (63.36 cM)区间, 其中一个位于346 cM处, 与Vr1-545的遗传距离为25.99 cM; 另一个位于361 cM处, 与Vr3-1177的遗传距离为22.37 cM, 遗传贡献率分别为67.82%、80.36%。
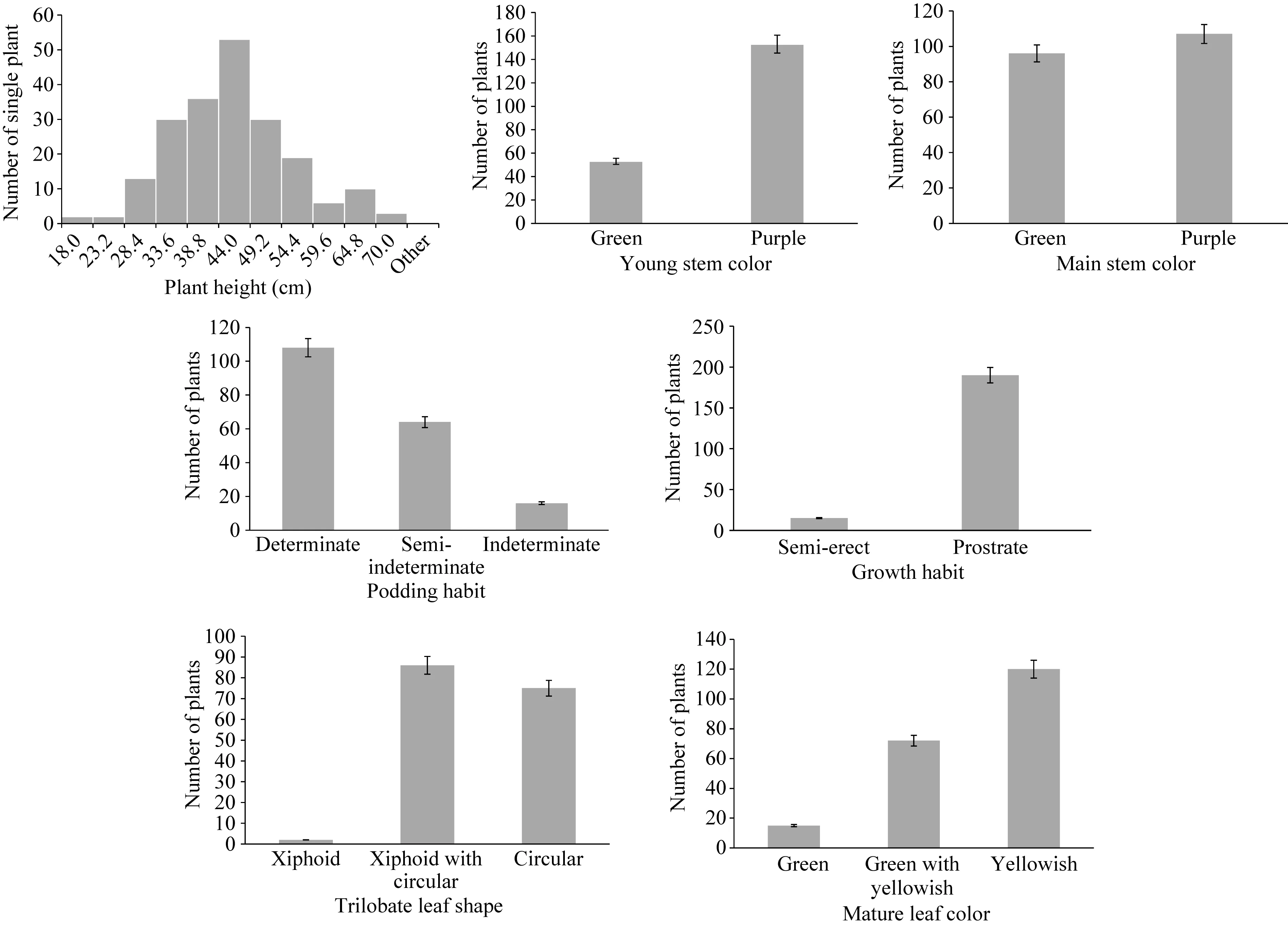
图1 F2群体中数量性状与质量性状的分布Fig. 1 Distribution of quantitative and quality traits in F2population
检测到 26个生长习性 QTL, 一个位于连锁群 1的Vr3-791–Vr3-303 (3.55 cM)区间, 与Vr3-303的遗传距离为1.58 cM, 遗传贡献率为58.32%; 16个位于连锁群2的Vr3-837–Vr2-440 (25.54 cM)、Vr2-440–Vr1-1509 (53.86 cM)区间, 遗传贡献率均为 99.51%; 8个位于连锁群 4的Vr3-1117–Vr3-1177 (98.33 cM)区间, 遗传贡献率均为99.51%; 1个位于连锁群10的Vr3-1246–Vr3-40 (17.87 cM)区间, 与Vr3-40的遗传距离为6.29 cM, 遗传贡献率均为99.51%。
检测到一个复叶叶形QTL, 位于连锁群4的Vr1-598~Vr1-2059 (67.79 cM)区间, 与 Vr1-2059的遗传距离为31.32 cM, 遗传贡献率为54.96%。
检测到4个成熟期叶色QTL, 2个位于连锁群2, 其中一个位于Vr2-1675–Vr3-107 (52.14 cM)区间, 与Vr3-107的遗传距离为 25.14 cM; 另一个位于 Vr2-440–Vr1-1509 (53.86 cM)区间, 与Vr1-1509的遗传距离为25.56 cM, 遗传贡献率分别为87.35%和69.06%; 2个位于连锁群4, 其中一个位于Vr1-598–Vr1-2059 (67.79 cM)区间, 与Vr1-2059的遗传距离为 32.32 cM; 另一个位于 Vr04-12148918–Vr2-1042 (45.49 cM)区间, 与Vr04-12148918的遗传距离为21.49 cM, 遗传贡献率分别为87.2%、84.3%。
3 讨论
3.1 绿豆遗传图谱的构建
遗传图是定位和挖掘基因的基本工具。绿豆现有遗传图的标记密度仍然很低, 并且只有少数基因被定位。早期图谱由 RFLP标记构建的 14个连锁群组成, 平均标记间距为9 cM[18], 另2个图谱由12个连锁群组[19]组成, 而基于RFLP标记的另一个连锁图谱只有9个连锁群[20]。Wang等[21]整合了一张含有560个标记物的11个连锁群遗传图谱, 连锁群的长度为 45.2~117.0 cM, 总覆盖度为 732.9 cM, 平均间隔在1.3 cM之间, 是比较饱和的遗传图谱。本研究所构建的新图谱大于钟敏[11]构建图谱的长度735.0 cM, 小于赵丹等[10]的 1831.8 cM, 且标记间距远大于Humphry等[22]和Wang等[21]的3.0 cM和1.3 cM, 图谱上的标记较少。其主要原因是试验所用的亲本都为栽培品种,与前人用的野生种、野生种加栽培种相比, 栽培种之间的遗传背景相似, 基因组变化较小, 所以筛选到的多态性引物比较少, 导致所绘制的连锁图谱长度比之前研究发表的图谱长, 标记间平均距离大。另外本文使用的是F2群体的208个家系, 前人研究所用材料为RIL群体, 对图谱的构建也会造成一定影响。

表3 绿豆7个产量相关性状的QTL及其遗传分析Table 3 QTLs for seven yield-related traits and their genetic analysis in F2population

图2 检测到的主效QTL在连锁群上的分布(LG)Fig. 2 Location of additive effects QTLs on linkage groupsGH: growth habit; MLC: mature leaf color; PHA: podding habit; TLS: trilobate leaf shape; MSC: main stem color; YSC: young stem color; PH: plant height.
3.2 绿豆农艺性状的QTL定位相关分析
本研究所构建图谱与赵丹等[10]、钟敏[11]加密连锁图谱的QTL定位结果相比, 将株高QTL定位在LG8, 与梅丽[23]的研究结果(第 2、第 8、第 9连锁群)基本一致, 对其他6个农艺性状都是首次进行QTL定位。且本研究中发现, 控制生长习性的QTL主要集中在第2、第4连锁群,且成簇出现。在第2连锁群Vr3-837~Vr1-1509之间79.4 cM, 检测到16个生长习性QTL, 且各个位点之间的距离为2~15 cM, 第4连锁群Vr3-1117~Vr3-1177之间98.33 cM, 检测到 8个生长习性 QTL, 且各个位点之间的距离为2~31 cM。在钟敏[11]的研究中也发现相同的现象, 影响荚长的QTL在第9连锁群的P3-334~P1-37有2个。复叶叶形(TLS) QTL被定位在LG4的Vr1-598~Vr1-2059之间,相距67.79 cM, 且Vr1-598属于第5染色体, Vr1-2059属于第 8染色体, 而 Jiao等[24]将叶瓣边缘基因(lma)定位在绿豆的Chr.3, 结果不一致。
对比已知的绿豆基因组染色体标记可知, LG2= Chr.9; LG3上43%的标记与LG5上全部标记对应于绿豆基因组的第1染色体上, 理论上LG3和LG5为同一条连锁群, 即LG3+ LG5=Chr.1; LG8与LG11的所有标记全都对应于绿豆基因组的第7染色体上, 理论上LG8和LG11为同一条连锁群, 即LG8+LG11=Chr.7; LG9上60%标记全都对应于绿豆基因组的第 11条染色体上,理论上LG9=Chr.11; LG4上由Chr.4、Chr.5和Chr.8的标记所组成, 理论上应该拆分为3条染色体, 即Chr.4、Chr.5和Chr.8。其余的连锁群上标记分布散乱, 无规律可循, 导致上述结果的原因可能是标记数量少、各连锁群上的标记区间过大。实验所观测的7个农艺性状主要集中在 LG1、LG2、LG4、LG8、LG10上, 其中 LG1最少, LG4最多; 其余连锁群没有检测到相关农艺性状的QTL。
今后的研究工作应注重构建新的 NIL群体用于连锁图谱分析, 增加结果的可靠性, 并开发新的标型记类, 提高引物多态性, 提高连锁图谱上的标记密度。
References
[1] Kang Y J, Kim S K, Kim M Y. Genome sequence of mungbean and insights into evolution within Vigna species. Nat Commun, 2014, 11: 1–7
[2] Cantarel B L, Korf I, Robb S M, Parra G, Ross E, Moore B, Holt C, Sánchez Alvarado A, Yandell M. MAKER: an easy-to-use annotation pipeline designed for emerging model organism genomes. Genome Res, 2008, 18: 188–196
[3] 赵金荣, 王晓玲, 白羊年. 豆科植物比较基因组学研究进展.见: 海南生物技术研究与发展研讨会论文集, 海南三亚, 2006. pp 65–75
Zhao J R, Wang X L, Bai Y N. Advances in Comparative Genomics of Leguminous Plants. In: Proceedings of Hainan Biotechnology Research and Development Symposium, Sanya, China, 2006. pp 65–75 (in Chinese with English abstract)
[4] Humphry M E, Magner T, McIntyre C L, Aitken E A B, Liu C J. Identification of a major locus conferring resistance to powdery mildew (Erysiphe polygoni DC) in mungbean (Vigna radiata L. Wilczek) by QTL analysis. Genome, 2003, 46: 738–744
[5] Reddy S K. Identification and inheritance of a new gene for powdery mildew resistance in mungbean (Vigna radiata L. Wilczek). Plant Breed, 2009, 128: 521–523
[6] Khajudparn P, Wongkaew S. Identification of genes for powdery mildew resistance in mungbean. J Life Sci, 2010, 4(8): 25
[7] Chaitieng B, Kaga A, Han O K, Wang X W, Wongkaew S, Laosuwan P, Tomooka N, Vaughan D A. Mapping a new source of resistance to powdery mildew in mungbean. Plant Breed, 2002, 121: 521–525
[8] Young N D, Danesh D, Menancio-Hautea D, Kumar L. Mapping oligogenic resistance to powdery mildew in mungbean with RFLPs. Theor Appl Genet, 1993, 87: 243–249
[9] 梅丽, 王素华, 王丽侠, 刘长友, 孙蕾, 徐宁. 重组近交系群体定位绿豆抗绿豆象基因. 作物学报, 2007, 33: 1601–1605
Mei L, Wang S H, Wang L X, Liu C Y, Sun L, Xu N. Mapping of genes resistant to Bruchid in mungbean using recombinant inbred lines population. Acta Agron Sin, 2007, 33: 1601–1605 (in Chinese with English abstract)
[10] 赵丹, 程须珍, 王丽侠, 王素华, 马燕玲. 绿豆遗传连锁图谱
的构建. 作物学报, 2010, 36: 932–939
Zhao D, Cheng X Z, Wang L X, Wang S H, Ma Y L. Integration of mungbean (Vigna radiata) genetic linkage map. Acta Agron Sin, 2010, 36: 932–939 (in Chinese with English abstract)
[11] 钟敏. 绿豆遗传连锁图谱的构建及抗豆象基因的精细定位.中国农业科学院硕士学位论文, 北京, 2012
Zhong M. Construction of Genetic Linkage Map and Fine Mapping of Bruchid-resistant Gene (Br1) of Mungbean. MS Thesis of Chinese Academy of Agricultural Sciences, Beijing, China, 2012 (in Chinese with English abstract)
[12] 吴传书, 王丽侠, 王素华, 陈红霖, 吴建新, 程须珍, 杨晓明.绿豆高密度分子遗传图谱的构建. 中国农业科学, 2014, 47: 2088–2098
Wu C S, Wang L X, Wang S H, Chen H L, Wu J X, Cheng X Z, Yang X M. Construction of a genetic linkage map in mungbean. Chin Agric Sci, 2014, 47: 2088–2098 (in Chinese with English abstract)
[13] Wang D L, Zhu J, Li Z K, Paterson A H. Mapping QTLs with epistatic effects and QTL × environment interactions by mixed linear model approaches. Theor Appl Genet, 1999, 99: 1255-1264
[14] Yang J, Zhu J. Predicting superior genotypes in multiple environments based on QTL effects. Theor Appl Genet, 2005, 110: 1268-1274
[15] Wang L X, Baidouri M E, Abernathy B, Chen H L, Wang S H, Cheng X Z. Distribution and analysis of SSR in mung bean (Vigna radiata L.) genome based on an SSR-enriched library. Mol Breed, 2015, 35: 25, DOI: 10.1007/s11032-015-0259-8
[16] Doyle J J, Doyle J L. A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem Bull, 1987, 19: 11–15
[17] 程须珍, 王素华, 王丽侠. 绿豆种质资源描述规范和数据标准.北京: 中国农业出版社, 2006. pp 3–38
Cheng X Z, Wang S H, Wang L X. Descriptors and Data Standard for Mungbean. Beijing: China Agriculture Press, 2006. 3–38 (in Chinese)
[18] Menancio-Hautea D, Fatokun C A, Kumar L, Danesh D, Young N D. Comparative genome analysis of mungbean (Vigna radiata L. Wilczek) and cowpea (V. unguiculata L. Walpers) using RFLP mapping data. Theor Appl Genet, 1993, 86: 797–810
[19] Lambrides D J, Lawn R J, Godwin I D, Manners J, Imrie B C. Two genetic linkage maps of mungbean using RFLP and RAPD markers. Aust J Agric Res, 2000, 51: 415–425
[20] Hautea D M, Legume I. Molecular mapping of drought resistance in mungbean [Vigna radiata (L.) Wilczek]: 1. Linkage map in mungbean using AFLP markers, J Bioteknologi Pertanian, 2002: 7: 17–24
[21] Wang L X, Wu C S. Construction of an integrated map and location of a bruchid resistance gene in mung bean. Crop J, 2016, 4: 360–366
[22] Humphry M E, Konduri V, Lambrides C J, Magner T, McIntyre C L, Aitken E A B, Liu C. Development of a mungbean (Vigna radiata) RFLP linkage map and its comparison with lablab (Lablab purpureus) reveals a high level of colinearity between the two genomes. Theor Appl Genet, 2002, 105: 160–166
[23] 梅丽. 绿豆抗豆象、种子硬实及其他重要农艺性状的QTL分析. 中国农业科学院硕士学位论文, 北京, 2007
Mei L. QTL Analysis of Bruchid Resistance, Seed Dormancy and Other Important Agronomic Traits in Mungbean. MS Thesis of Chinese Academy of Agricultural Sciences, Beijing, China, 2007 (in Chinese with English abstract)
[24] Jiao K Y, Li X, Guo W X, Yuan X X, Cui X Y, Chen X. Genome re-sequencing of two accessions and fine mapping the locus of lobed leaflet margins in mungbean. Mol Breed, 2016, 36: 128
Construction of Genetic Map and Identification of QTLs Related to Agronomic Traits in Mung Bean
WANG Jian-Hua1,2,3, ZHANG Yao-Wen3, CHENG Xu-Zhen2,*, and WANG Li-Xia2,*
1Shanxi University, Taiyuan 030006, China;2Institute of Crop Science, Chinese Academy of Agricultural Sciences, Beijing 100081, China;3Institute of Crop Science, Shanxi Academy of Agricultural Sciences, Taiyuan 030031, China
Two hundreds and eight individuals of F2population, derived from a cross between two mung bean genotypes (Huaye 1 and Zijing 1) were used to construct genetic map, and to identify QTLs related to important agronomic traits. This genetic map contained 11 linkage groups with a total length of 1457.47 cM and an average interval of 15.34 cM. QTLs mapping was conducted for plant height, young stem color, main stem color, growth habit, podding habit, trilobate leaf shape and mature leaf color using composite interval mapping method. Only one QTL for each trait was detected including plant height, young stem color, main stem color and trilobate leaf shape, and with a contribution ranging from 8.49% to 66.64%. Three QTLs with high contribution rates from 60.32% to 80.36% were identified for the trait of pod habit in mung bean. Four QTLs related to mature leaf color showed at contribution rate from 69.06% to 87.35%. There were 26 QTLs related to growth habit, the most of the tested QTLs, with a contribution rate each from 58.32% to 99.51%. The present QTLs for seven agronomic traits distributed on LG1, LG2, LG4, LG8, and LG10, respectively, could be used in molecular breeding based on marker-assisted selection in mung bean, and also lay a foundation for further study of the inheritance of these traits.
Mung bean; Genetic linkage map; SSR markers; Composite interval mapping (CIM); QTL; Contribution rate
(
): 2016-12-11; Accepted(接受日期): 2017-03-02; Published online(网络出版日期): 2017-03-19.
10.3724/SP.J.1006.2017.01096
本研究由国家现代农业产业技术体系建设专项(CARS-09)和中国农业科学院农业科技创新工程资助。
This study was supported by the China Agriculture Research System (CARS-09) and the Agricultural Science and Technology Innovation Program of CAAS。
*通讯作者(Corresponding authors): 程须珍, E-mail: chengxuzhen@caas.cn, 王丽侠, E-mail: wanglixia03@caas.cn, Tel: 010-62180535
联系方式: E-mail: 986254540@qq.com
URL: http://kns.cnki.net/kcms/detail/11.1809.S.20170319.1749.002.html
