碳纳米管超晶格结构吸附Fe原子的电子机理*
2017-07-19杨忠华李荣德曲迎东沈阳工业大学建筑与土木工程学院材料科学与工程学院沈阳110870
杨忠华, 李荣德, 曲迎东(沈阳工业大学 a. 建筑与土木工程学院, . 材料科学与工程学院, 沈阳 110870)
碳纳米管超晶格结构吸附Fe原子的电子机理*
杨忠华a,b, 李荣德b, 曲迎东b
(沈阳工业大学 a. 建筑与土木工程学院, b. 材料科学与工程学院, 沈阳 110870)
为了提高碳纳米管与Fe基体之间的润湿性,构造出N掺杂有限长碳纳米管超晶格结构.第一性原理能量计算结果表明,新型超晶格结构的埋置能正向升高,结构稳定性降低,但可以显著提高外壁对Fe原子的吸附能力.差分电荷密度结果表明,掺杂体系中N原子与邻近C原子间的π键出现了畸变,使得N原子易与Fe原子发生结合.布居数和电荷转移情况表明,N原子的掺入导致Fe原子失电子能力降低,但Fe—N间共价键强度提高.超晶格结构在一定的扭转和剪切变形下仍能保持对Fe原子的吸附能力.
碳纳米管; 超晶格结构; N原子掺杂; Fe原子吸附; 电子机理; 第一性原理; 剪切变形; 扭转变形
随着经济发展与科技进步,人们对材料的性能与多样化的要求日趋提高.目前,应用最为广泛的合金材料已经不能满足实际应用的需要,因而对传统材料进行改进制备出新型材料势在必行.球墨铸铁是二十世纪五十年代发展起来的一种铸铁材料,以其优异的综合力学性能和相对低廉的成本已被大范围应用.铁素体球墨铸铁具有较高的伸长率,但是硬度和强度较低,因而其应用范围受到一定限制.单壁碳纳米管是由单层石墨卷曲形成的具有较高对称性的空心圆柱体,且自1991年被发现以来一直是相关领域的研究热点[1].由于具有优异的力学性能(高强度、高韧性和较好的化学稳定性)和较大的比表面积,碳纳米管已经成为增强金属基复合材料的理想候选材料.但在制备过程中由于碳纳米管管壁化学活性较低,很难与金属基体润湿形成牢固的结合.张文忠[2]和徐强[3]等利用修饰碳纳米管制备得到了增强Cu、Fe基复合材料,但在研究中发现,制约该复合材料发展的主要原因是碳纳米管与铁液之间不发生浸润,因而碳纳米管难于进入铁基金属熔体中,这在物理角度表现为碳纳米管与Fe原子间的吸附问题.近年来,随着碳纳米管制备技术的长足进步和原子显微镜在纳米材料领域的广泛应用,已经实现了对碳纳米管各原子的定位操控[4-5].将碳纳米管进行表面改性处理(如包覆、掺杂等),可以获得优于纯碳纳米管的物理化学性质[6].
半导体材料中的超晶格结构由交替生长的半导体薄层组成.目前,在碳纳米管相关研究工作中超晶格结构的概念鲜见报道.本文首次构造出N掺杂有限长碳纳米管结构,即在该结构中掺杂层与石墨层交替出现,本文借用超晶格结构概念,称其为N掺杂碳纳米管超晶格结构.运用第一性原理研究了该新型超晶格结构对Fe原子吸附性能的改善作用,并通过外加变形作用(如扭转、剪切变形),考察了Fe原子吸附能力的稳定性,以期为碳纳米管增强Fe基复合材料的深入研究与应用提供科学依据.
1 物理模型与计算方法
碳纳米管是由石墨烯卷曲形成的无缝圆筒,圆筒两端在“共振稳定化能”的驱动作用下形成的笼状富勒烯物质可以通过机械球磨或化学腐蚀等方法予以去除.由于开口后的碳纳米管物理化学活性增大而常被选作复合材料的增强相,故本文以两端开口且具有金属性质的(5,5)扶手椅型单壁碳纳米管为研究对象.由洪特规则可知,当等价轨道呈全充满、半充满或全空状态时,其电子排布较为稳定.在碳纳米管中C原子核外电子构型为1s22s22p2,最外层的4个电子通过C—C原子间的sp2轨道杂化形成3个σ键和1个π键,从而获得稳定结构.
有限长碳纳米管的原子结构如图1所示.在该结构中需要沿x和y轴向方向添加1 nm的隔离层,从而避免能量计算时各周期结构间的相互影响.图1中大球表示C原子,小球表示H原子.
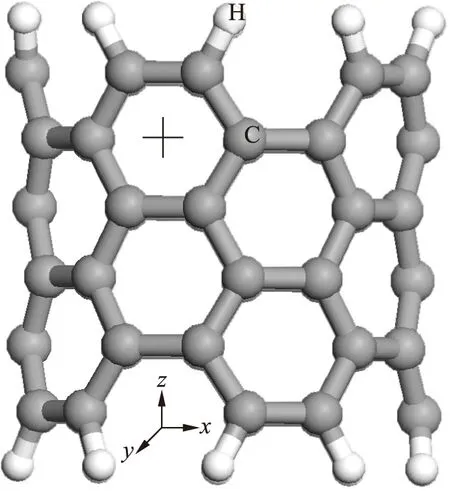
图1 有限长碳纳米管的原子结构Fig.1 Atom structure of CNT with finite length
图2为N掺杂有限长碳纳米管的原子结构.与C原子不同,N原子核外电子构型为1s22s22p3,2个σ键和1个π键即可饱和核外电子轨道,故由N原子替换碳纳米管上下端口处的C原子形成的超晶格结构中不存在未饱和的悬挂键.图2中大球表示N原子,小球表示C原子.
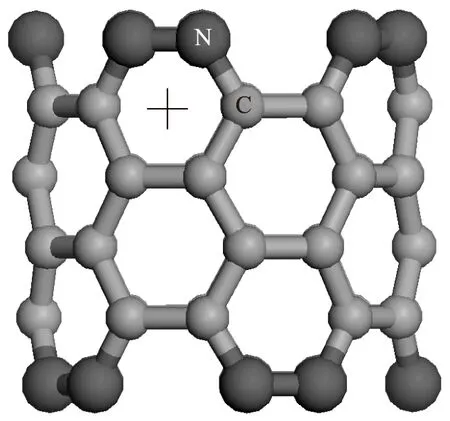
图2 N掺杂有限长碳纳米管的原子结构Fig.2 Atom structure of N-doped CNT with finite length
Philip等[7]总结归纳了近年来利用密度泛函理论在预测材料结构参数方面取得的成果后指出,CASTEP程序能够获得切实可信的精度.本文中交换关联参数采用由广义近似梯度GGA修正的PW91泛函进行计算,同时采用倒易空间Ultrasoft赝势,平面波截断能为340 eV,采用Monkhorst-Pack生成方法选取布里渊区K点,且K点网格设定为1×1×10.综合考虑到计算成本与精度,设定单个原子的自洽允许误差为10-6eV,键能误差为10-5eV,最大应力收敛精度为0.05 GPa,最大位移收敛精度为10-4nm.当纯(5,5)碳纳米管模型完全弛豫后,C—C原子间键长介于0.137~0.144 nm之间,该计算值与文献[8]所得到的C—C键长(0.138~0.145 nm)相吻合,相对误差小于1%.由此可知,本文计算参数设置合理,结果可信.
2 计算结果与分析
2.1 埋置能与吸附能计算
当N原子有规律地取代单壁碳纳米管的C原子环时,其对环境能量的改变可以通过计算埋置能的方式进行表征.埋置能的计算公式为
EI=ECNT+N-ECNT-nEN+nEC
(1)
式中:ECNT+N为N掺杂碳纳米管中C原子环的系统总能量;ECNT为去除悬挂键后纯碳纳米管的系统总能量;n为系统中所替换原子的数目;EN和EC分别为N、C原子在孤立状态下的原子总能量.
埋置能为正值表明异质原子的掺入使得周围环境能量提高,并使结构稳定性降低;反之,结构稳定性提高.将N、C原子放入边长为1 nm的立方体晶盒中,弛豫得到N、C原子在孤立状态下的原子总能量分别为-262.696与-146.025 eV.埋置能计算结果表明,平均每个N原子可以使周围环境能量提高5.071 eV,使得结构稳定性降低.由此可见,通过原子操控技术构造N掺杂有限长碳纳米管超晶格结构时需要额外能量的输入.
利用第一性原理研究了构造出的新型超晶格结构外壁对Fe原子吸附性能的影响.Yagi等[9]指出,当Fe原子吸附在石墨烯C原子六元环中心(洞位)时更具稳定性.因而将Fe原子放置于有限长碳纳米管C原子的六元环洞位,即图1中十字线位置处.同理,在N掺杂碳纳米管超晶格结构中Fe原子吸附在图2中的十字线位置处.调整Fe原子与纳米结构外壁的距离,使得洞位处的Fe原子与周围6个原子相距0.21 nm(Fe—C化合物的键长约为0.21 nm).此时吸附能的计算公式为
Ea=ECNT+EFe-ECNT+Fe
(2)
式中:EFe为吸附单个Fe原子的能量;ECNT+Fe为吸附Fe原子后单壁碳纳米管的系统总能量.
计算结果表明,有限长碳纳米管外壁对Fe原子的吸附能为0.703 eV.强碳化物形成元素Ti、V、Nb、Mn、Cr、W和Mo在(8,0)碳纳米管外壁的吸附能分别为2.9、3.2、3.9、3.4、3.7、3.4和4.6 eV[10].可见,有限长碳纳米管外壁对Fe原子的吸附能较低,因而很难与Fe基体间形成牢固的界面.N掺杂碳纳米管外壁对Fe原子的吸附能为1.811 eV,相比纯碳纳米管外壁对Fe原子的吸附能呈现出了较大幅度提高,因而可以改善碳纳米管增强Fe基复合材料界面的黏附结合性.
2.2 差分电荷密度、布居数和电荷转移分析
在进行能量计算的同时,本文也分析了纯碳纳米管和N掺杂碳纳米管结构的差分电荷密度,具体结果如图3所示.
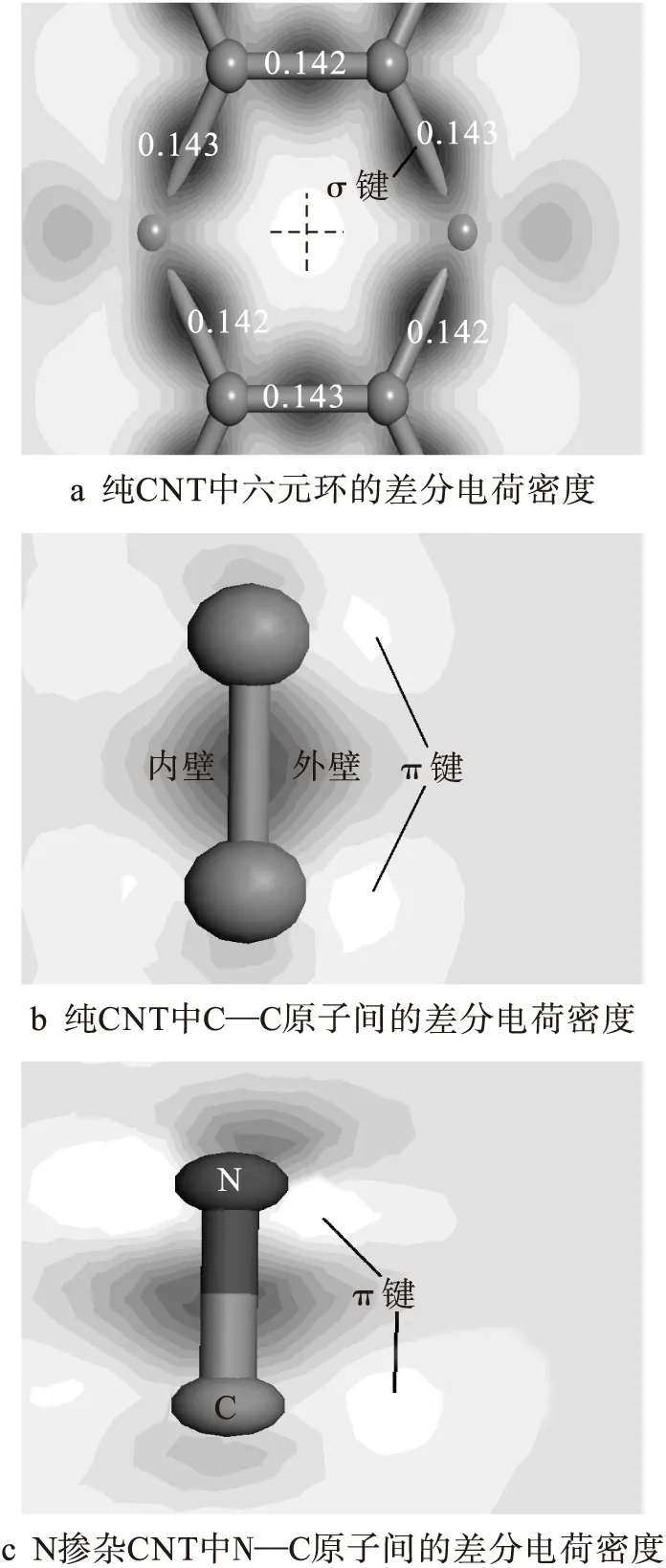
图3 差分电荷密度Fig.3 Difference charge density
由纯碳纳米管和N掺杂碳纳米管结构的差分电荷密度计算结果可知,两种结构的差分电荷密度跨度区间从-1.63×102e/nm3(白色区域)变化至8.52×102e/nm3(黑色区域),其中e为电荷量的单位.图3a为未掺杂碳纳米管六元环的差分电荷密度,其中六元环上的数字代表键长(单位:nm).由图3a可见,C—C原子间的σ键由于发生弯曲使其键长具有一定的分散度,但其键长始终处于0.142~0.143 nm范围内.共用电子对形成的差分电荷密度集中区位于C—C原子间中心,从而使体系具有良好的电中性.六元环中心的空白差分电荷密度区大体呈圆形,符合碳纳米管差分电荷密度分布特点.图3b、c分别为掺杂前后碳纳米管外壁剖面的差分电荷密度.对比图3b、c后发现,N—C原子间差分电荷密度降低,σ键强度减弱.在N原子附近出现了多电子区域,致使N—C原子间存在离子键.计算纯碳纳米管和掺杂碳纳米管体系的布居数和电荷转移情况后发现,N—C原子间的布居数0.89小于C—C原子间的布居数0.97,但在纯碳纳米管中C—C原子间无电荷转移,而N—C原子间电荷转移了0.16 e,可见N原子的掺入使得N—C原子间的共价键相比C—C原子间减弱,而离子键增强.由图3c可见,N原子的掺入破坏了碳纳米管结构的电中性,不仅N原子附近呈现多电子状态,而且N原子与邻近C原子间的π键也发生了畸变.众所周知,π键的形貌决定了材料的物理化学活性.π键的畸变使得掺杂碳纳米管体系易于与金属原子发生反应结合.两种吸附体系的布居数及电荷转移数计算结果表明,在纯碳纳米管吸附体系中Fe原子与C原子间的布居数为负值,表明原子间无共价键存在,同时Fe原子失去电荷1.13 e,周围六元环C原子平均得到电荷-0.17 e.在N掺杂碳纳米管吸附体系中Fe原子失去电荷1.06 e,但Fe—N原子间的布居数为0.22,此时存在一定的共价键.因此,在综合作用下N掺杂碳纳米管超晶格结构对Fe原子的吸附能力得到了较大提升.
2.3 吸附体系态密度分析
为了分析新型超晶格结构对Fe原子吸附性能的影响机理,计算并分析了N掺杂碳纳米管与纯碳纳米管吸附体系的态密度,具体结果如图4所示.
图4a为纯(5,5)碳纳米管和N掺杂(5,5)碳纳米管吸附体系的总态密度.由图4a可见,两种吸附体系的总态密度在费米能级(能量为0 eV)附近发生根本转变,N掺杂碳纳米管体系的总态密度位于纯碳纳米管体系之上.费米能级附近的态密度对物质活度具有很大影响,态密度较大则其活度较高,态密度较小则其活度较小.由于N掺杂碳纳米管体系的活度升高,此时的碳纳米管更易与外来物质发生反应,因而对Fe原子也更具吸附性,这与N—C原子间的π键畸变分析结论一致.图4b为N掺杂碳纳米管吸附体系中Fe3d轨道与N2p轨道的分波态密度.由图4b可见,在能量小于0 eV的低能区Fe3d轨道与N2p轨道间共振区较多,Fe—N原子间化学结合作用明显.图4c为纯碳纳米管吸附体系中Fe3d轨道与C2p轨道的分波态密度.由图4c可见,两种轨道之间无明显共振区,且Fe—C原子间的相互作用较弱.图4d为两种吸附体系中Fe3d轨道的分波态密度.由图4d可见,N掺杂碳纳米管吸附体系中Fe3d轨道的分波态密度在费米能级附近相对较低,活度较弱,表明经过新型结构吸附后Fe原子的物理化学活性降低.因此,N掺杂使得碳纳米管活性提高,Fe—N原子间易于发生化学结合作用,因而结构对Fe原子更具吸附性,这一结论与吸附能计算结果一致.
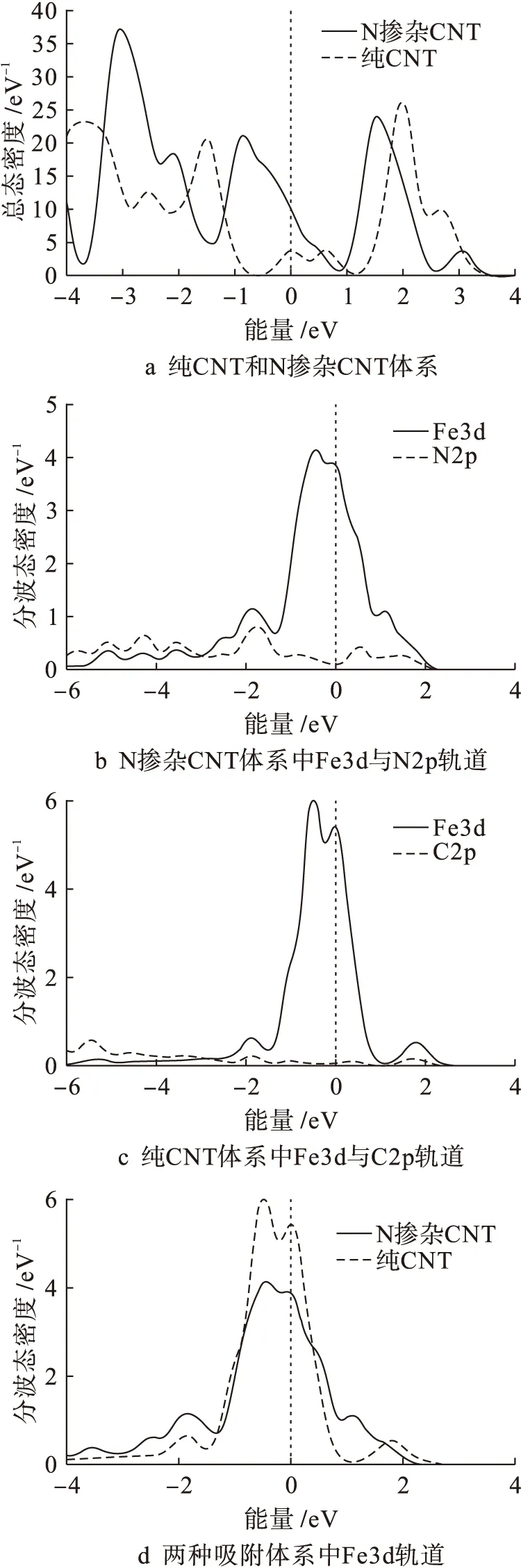
图4 态密度和分波态密度Fig.4 Density of states and partial density of states
2.4 剪切与扭转变形对Fe原子吸附能的影响
N掺杂有限长碳纳米管超晶格结构可以提高外壁对Fe原子的吸附性能,因而经过掺杂后的碳纳米管可以作为复合材料的增强相并与基体间形成结合良好的界面.在实际应用中碳纳米管作为准一维纳米材料主要受到拉伸、压缩变形作用,但随机弥散分布在基体中的碳纳米管的轴线可能与复合材料的轴线存在一定角度,使得外壁呈现复杂的应力状态,从而影响增强体与基体界面间的黏附性.因而需要讨论变形作用对Fe原子吸附性能的影响.
图5为N掺杂有限长碳纳米管的扭转变形示意图.有限长碳纳米管包含6层C原子环,将6层C环由下至上沿扭转方向施加不大于1°的扭转变形量,并将第1和第6层C环固定,即相当于对超晶格结构持续施加扭转变形,此时结构外壁与Fe原子间出现排斥力,且相应吸附能为-1.667 eV.与纯碳纳米管静态体系的吸附能0.703 eV相比,吸附能变化率高达337%.可见,微小扭转变形将严重恶化纯碳纳米管对Fe原子的吸附能力,且在碳纳米管增强Fe基复合材料中将出现增强相与基体间发生脱粘的现象.同理,对N掺杂有限长碳纳米管超晶格结构施加扭转变形,当扭转变形角度为1、2、3、4和5°时,计算得到的结构外壁对Fe原子的吸附能分别为0.901、0.541、0.241、0和-0.529 eV.由于此时N掺杂有限长碳纳米管静态体系的吸附能为1.811 eV,因而扭转变形角度为1°时的吸附能与之相比其变化率为50.2%.由此可见,N掺杂碳纳米管超晶格结构对扭转变形恶化吸附能的现象具有一定的抗力.
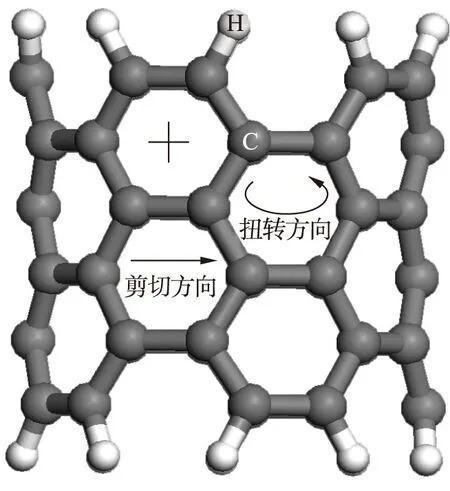
图5 N掺杂有限长碳纳米管的扭转变形示意图Fig.5 Schematic diagram of torsional deformation of N-doped CNT with finite length
此外,当分析纯碳纳米管结构在剪切变形作用下对Fe原子吸附性能的影响时,剪切变形的施加方法与扭转变形的施加方法相似,即将由下至上的6层C环沿剪切方向施加不大于0.01 nm的剪切变形量,并将第1和第6层C环固定,这相当于对结构持续施加剪切变形.此时,纯碳纳米管外壁对Fe原子的吸附能为-1.707 eV,与静态体系吸附能0.703 eV相比,吸附能变化率为343%.同理,对N掺杂碳纳米管结构施加相同的剪切变形量,此时结构外壁对Fe原子的吸附能为0.721 eV,与相应静态体系的吸附能相比其变化率为60.2%.因此,在剪切变形作用下N掺杂碳纳米管体系对吸附能的恶化仍然具有一定的抗力.当剪切变形量为0.015、0.02和0.025 nm时,N掺杂碳纳米管外壁对Fe原子的吸附能分别为0.501、0.371和0.201 eV,此时N掺杂碳纳米管结构仍然对Fe原子具有吸附性.
综上所述,在一定的变形作用下N掺杂有限长碳纳米管超晶格结构对吸附能的恶化现象具有一定抗力,因而碳纳米管增强Fe基复合材料在实际使用中能够保持界面稳定性,这具有十分重要的意义.
3 结 论
本文采用基于密度泛函理论的CASTEP程序分析了N掺杂有限长碳纳米管超晶格结构的能量参数、电子结构以及在变形作用下结构外壁对Fe原子吸附能力的变化规律.结果表明,N掺杂碳纳米管结构的埋置能正向升高,结构稳定性降低,结构中N原子周围呈现多电子状态,N—C原子间的离子键加强,而共价性减弱.N掺杂碳纳米管结构可以显著提高外壁对Fe原子的吸附能力,这是由于N原子与邻近的C原子间的π键出现了畸变,使得结构的物理化学活性升高,同时Fe—N原子间轨道共振区增多,使得Fe—N原子间化学结合作用明显.在扭转、剪切变形作用下,N掺杂结构对吸附能的恶化具有一定的抗力,在一定的变形作用下N掺杂碳纳米管结构可以仍然保持对Fe原子的吸附性能,这对复合材料的实际应用具有重要意义.
[1]刘贵立,姜艳,谢萌.碳纳米管超晶格结构的第一性原理 [J].沈阳工业大学学报,2015,37(3):289-293.
(LIU Gui-li,JIANG Yan,XIE Meng.First principle for carbon nanotube super lattice structure [J].Journal of Shenyang University of Technology,2015,37(3):289-293.)
[2]张文忠,蔡晓兰,周蕾,等.CNTs修饰及其增强Cu基复合材料的研究 [J].材料工程,2016,44(12):1-6.
(ZHANG Wen-zhong,CAI Xiao-lan,ZHOU Lei,et al.CNTs modified and enhanced Cu matrix composites [J].Journal of Materials Engineering,2016,44(12):1-6.)
[3]徐强,曾效舒,徐春水,等.碳纳米管/球墨铸铁复合材料制备及组织性能的研究 [J].铸造技术,2010,31(2):165-168.
(XU Qiang,ZENG Xiao-shu,XU Chun-shui,et al.Study on the hardness and microstructure of carbon nanotubes/ductile cast-iron composite fabricated by lost foam casting [J].Foundry Technology,2010,31(2):165-168.)
[4]吴森.基于AFM的一维纳米材料操纵及力学特性测试技术 [D].天津:天津大学,2011.
(WU Sen.AFM based manipulation and mechanical properties measurement of one-dimensional nanomaterials [D].Tianjin:Tianjin University,2011.)
[5]陈清,魏贤龙.在扫描电子显微镜中原位操纵、加工和测量纳米结构 [J].电子显微镜学报,2011,30(6):473-482.
(CHEN Qing,WEI Xian-long.In-situ manipulating fabricating and measuring nanostructures inside SEM [J].Journal of Chinese Electron Microscopy Society,2011,30(6):473-482.)
[6]赵冬梅,李振伟,刘领弟,等.石墨烯/碳纳米管复合材料的制备及应用进展 [J].化学学报,2014,72(2):185-200.
(ZHAO Dong-mei,LI Zhen-wei,LIU Ling-di,et al.Progress of preparation and application of grapheme/carbon nanotube composite materials [J].Acta Chimica Sinica,2014,72(2):185-200.)
[7]Philip J H,Keith R,Matt I J P,et al.Density functional theory in the solid state [J].Philosophical Transactions of the Royal Society A,2014,372:1-26.
[8]刘贵立,宋媛媛,姜艳,等.拉压变形对B(N)掺杂碳纳米管Al吸附性能的影响 [J].沈阳工业大学学报,2016,38(4):391-396.
(LIU Gui-li,SONG Yuan-yuan,JIANG Yan,et al.Effect of tension and compression deformation on adsorption properties between Al and carbon nanotubes with B(N) doping [J].Journal of Shenyang University of Technology,2016,38(4):391-396.)
[9]Yagi Y,Briere T M,Sluiter M H F,et al.Stable geometries and magnetic properties of single-walled carbon nanotubes doped with 3d transition metals:a first-principles study [J].Physical Review B,2004,69(7):1-8.
[10]Makita T,Doi K,Nakamura K,et al.Structures and electronic properties of aluminum nanowires [J].Journal of Chemical Physics,2003,119(1):538-546.
(责任编辑:尹淑英 英文审校:尹淑英)
Electronic mechanism of Fe atom adsorbing for CNT supperlattice structure
YANG Zhong-huaa,b, LI Rong-deb, QU Ying-dongb
(a. School of Architecture and Civil Engineering, b. School of Materials Science and Engineering, Shenyang University of Technology, Shenyang 110870, China)
In order to improve the wettability between carbon nanotube (CNT) and Fe substrate, a N-doped CNT supperlattice structure with finite length was constructed. The energy calculation results based on the first principle show that the embedding energy of novel superlattice structure positively increases, the structure stability decreases, but the Fe atom adsorbing ability of outside wall gets significantly improved. The difference charge density indicates that in the doping system, the π bond connecting N atom and neighboring C atom has distorted, which allows N atom to bond with Fe atom easily. Both population and charge transfer situation show that the electron loss ability of Fe atom decreases due to the doping of N atom, but the intensity of Fe—N covalent bond increases. The superlattice structure can hold the ability to adsorb Fe atom under some extent of torsional and shear deformation.
carbon nanotube (CNT); superlattice structure; N atom doping; Fe atom adsorbing; electronic mechanism; first principle; shear deformation; torsional deformation
2016-08-24.
国家自然科学基金资助项目(51274142).
杨忠华(1985-),男,辽宁大连人,工程师,博士生,主要从事材料晶体结构等方面的研究.
10.7688/j.issn.1000-1646.2017.04.05
TG 113
A
1000-1646(2017)04-0383-06
*本文已于2017-06-21 21∶19在中国知网优先数字出版. 网络出版地址: http:∥www.cnki.net/kcms/detail/21.1189.T.20170621.2119.006.html
