MLVA用于新疆部分地区结核分枝杆菌基因分型的初步研究
2017-07-18龚新记李月华姚丽丹阿依努尔莫合买提刘年强王乐王晶
龚新记 李月华 姚丽丹 阿依努尔·莫合买提 刘年强 王乐 王晶
MLVA用于新疆部分地区结核分枝杆菌基因分型的初步研究
龚新记1李月华2姚丽丹3阿依努尔·莫合买提2刘年强2王乐2王晶4
目的探讨结核菌株进行多位点可变数目串联重复序列分析(the multiple locus VNTR analysis, MLVA)。方法选择2010年至2011年新疆维吾尔自治区第五次结核病流行病学抽样调查资料,采用经典24位点方法进行基因分型。并采用BioNumerics5.0数据库进行基因聚类分析。将结核分枝杆菌原始株,取一菌环溶于400 μl TE中悬菌,80 ℃ 1 h灭活,12 000 r/min离心10 min,弃上清,600 μl TE重新悬菌,进行多位点串联重复序列分析。结果新疆99株结核分枝杆菌分为2个基因群:分别为基因群Ⅰ和基因群Ⅱ,基因群Ⅰ66株(66.7%),基因株Ⅱ33株(33.3%);基因群Ⅰ是北京家族,基因群Ⅰ的66株结核分枝杆菌有65种不同的基因型,有2株结核分枝杆菌属于同一簇,成簇率为1.5%,基因群II33株结核分枝杆菌菌株的MLVA图谱不同,成簇率为0。结论新疆结核分枝杆菌菌株存在明显的基因多态性,以北京基因型菌株为主,同时还存在一定比例的非北京基因型,应加强对主要流行菌株流行的监控及管理。
结核病; 结核分枝杆菌; MLVA; 基因多态性; 新疆地区
结核病(tuberculosis, TB)是由结核分枝杆菌引起的慢性传染病,是目前世界范围内危害最严重的传染病之一[1-4]。结核病是单一致病菌(结核分枝杆菌)感染导致病死率最高的疾病之一,随着抗结核药物的研究发展,结核发病率和病死率曾经在一段时期内大幅度下降,但20世纪80年代后,由于艾滋病的流行、结核分枝杆菌耐药菌株出现,尤其是耐多药结核菌株的产生,使全球结核病发病率和病死率再次呈现上升趋势[5-7]。2010年至2011年新疆维吾尔自治区第五次结核病流行病学抽样调查显示全区活动性、涂阳和菌阳肺结核标化患病率分别为1 526.12/10万、196.43/10万和433.44/10万,表明新疆结核疫情仍然不容乐观[8]。为此,本研究旨在前期研究的基础上,对疑似结核者进行痰涂片及痰培养,并对获得菌株进行多位点可变数目串联重复序列分析(the multiple locus VNTR analysis, MLVA),采用经典24位点方法进行基因分型,并采用BioNumerics5.0数据库进行基因聚类分析,旨在了解新疆部分地区结核分枝杆菌的基因型分布及其流行情况,探讨其在今后结核病控制中的应用价值[9-14]。
材料与方法
一、实验材料
结核分枝杆菌标准菌株、BCG和DNA marker结核分枝杆菌标准菌株H37Rv为香港跨国参比实验室提供。BCG由传染病预防控制国家重点实验室/中国疾病预防控制中心结核病参比实验室提供。100bp DNA Marker购自华美生物工程公司。菌株来自2011年结核病流行病学抽样调查中筛选的1 571例可疑肺结核者的痰液。其中痰涂片阳性50例,痰培养阳性103例,其中4例为非结核分枝杆菌,对其余 99例进行提取DNA进行基因分型分析。Taq酶购自天根生物工程公司;dNTP购自上海生物工程公司。Gel Compar软件4.0版本,BioNumerics软件3.0版本。实验中引物合成委托北京赛百盛基因技术公司进行。
二、研究方法
1. 结核分枝杆菌DNA的制备:将结核分枝杆菌原始株,取一菌环溶于400 μl TE中悬菌,80 ℃1 h灭活,以离心半径8 cm,12 000 r/min离心10 min,弃上清,600 μl TE重新悬菌。再金属浴95 ℃15 min,以离心半径8 cm,12 000 r/min离心10 min,取上清即为模板,-20 ℃保存备用。
2. 多位点串联重复序列分析:根据文献报道和细菌基因数据库,筛选24个串联重复基因位点,见表1。
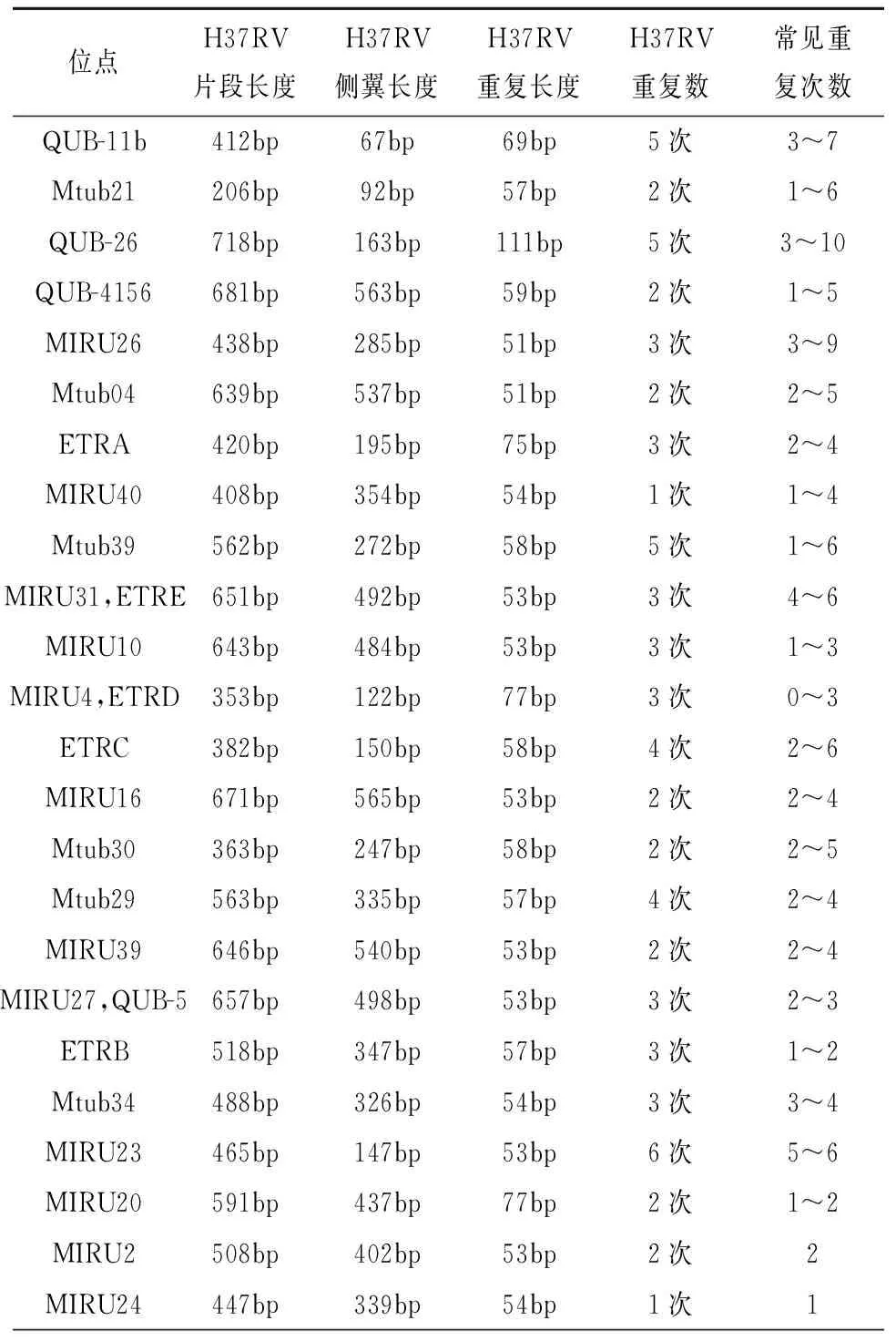
表1 VNTR 24经典位点H37Rv长度与重复次数
3. PCR反应体系:采用23 μl反应体系:含上下游引物(5 μmol/L)各0.5 μl,DNA模版5 μl,MIX 10 μl,DMSO 2 μl,去离子水5 μl补充至反应体系。引物序列见表2。
4. 反应条件:预变性95 ℃ 15 min,35个循环:95 ℃ 1 min、59 ℃ 1 min、72 ℃ 1.5 min后,72 ℃延伸10 min。
5. 方法分析:将菌株经PCR扩增后的凝胶电泳图片与标准分子量Marker对比,结合不同位点重复片段大小以及扩增产物侧翼大小,计算其重复次数,再用BioNumerics 5.0数据库软件进行聚类分析,将实验菌株分成不同的家族。
结 果
每次试验设立的阳性对照菌株H37Rv DNA在同一VNTR位点的扩增片段大小相同。从检测标本中抽取20株菌株,每株菌不同VNTR位点重复检测3次,每次DNA扩增片段大小完全相符。
本试验共选取24个VNTR基因位点对新疆99株结核分枝杆菌进行基因分型,结果显示不同菌株在MIRU不同位点的DNA图谱呈现多态性[15]。
采用最小生成树分型聚类分析[16-17],新疆99株结核分枝杆菌可分为2个基因群(分别为基因群Ⅰ和基因群Ⅱ),其中基因群Ⅰ所占比例较大,包括66株,占66.7%,基因群Ⅱ包括33株,占33.3%,经与Spoligotyping[18-20]结果综合对比分析,基因群Ⅰ是北京家族。此外,基因群Ⅰ的66株结核分枝杆菌包括65种不同的基因型,其中有2株结核分枝杆菌属于同一簇,成簇率为1.5%;而基因群Ⅱ的33株结核分枝杆菌菌株的MLVA图谱均不相同,成簇率为0%。
讨 论
多位点可变数量串联重复序列分析是通过基因组中可变数量串联重复序列(VNTR)的特征来实现分型的分子分型技术,由于该方法具有特异、敏感、简便、快速、分辨力强、分型效率高和重复性好等特点,可有效地对结核分枝杆菌进行基因分型,适合于所有结核菌分离株,成为结核流行地区的一线分型方法[21-23]。
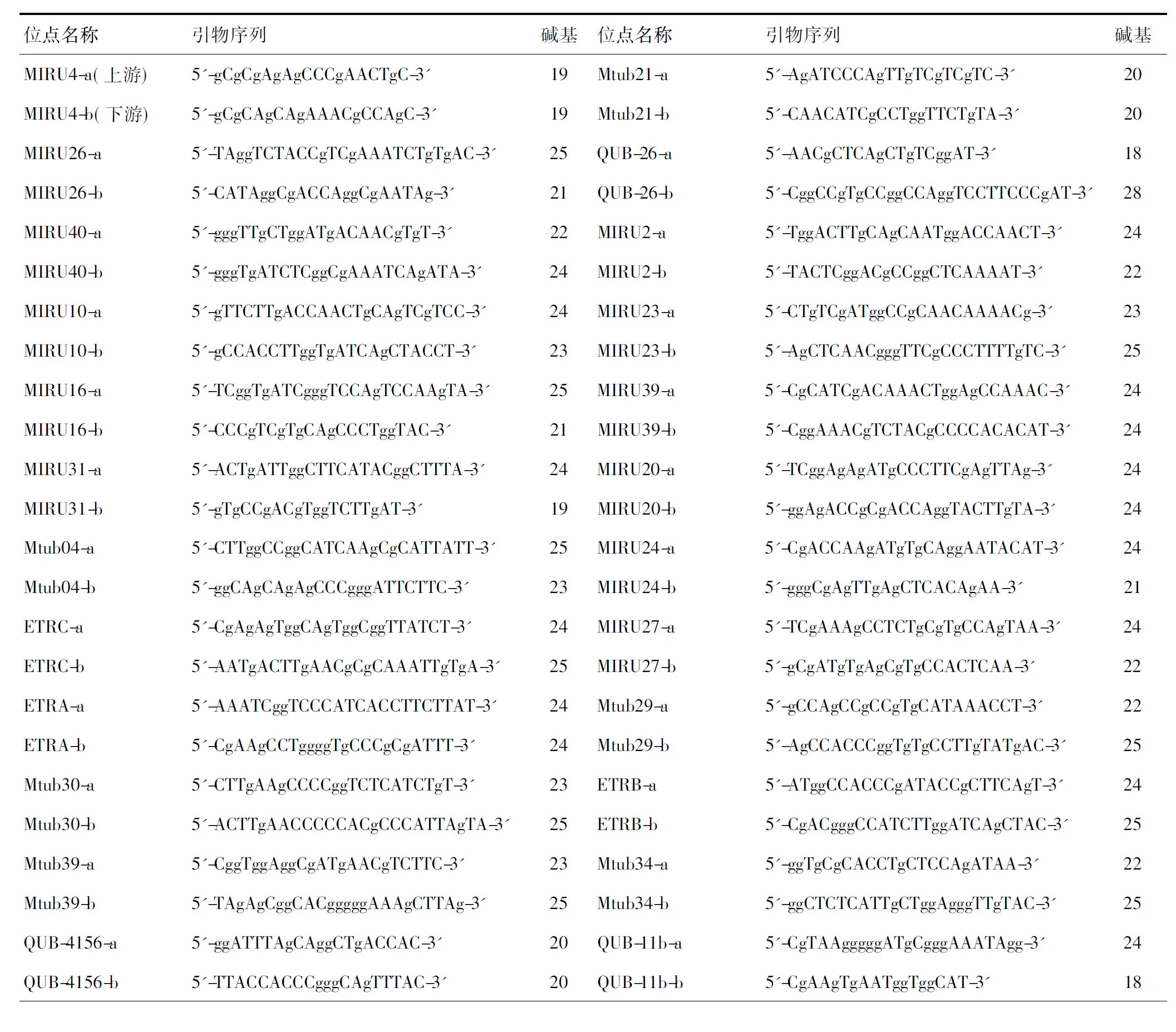
表2 24经典位点引物序列
在2011年结核病流行病学抽样调查中,对有可疑症状和胸片疑似肺结核者1571人进行痰培养工作,其中痰涂片阳性50例,阳性率为3.18%(高于全国196.43/10万),痰培养阳性103例,阳性率为6.56%(高于全国433.44/10万),说明新疆仍是全国结核的高发地区。新疆2010-2011年结核病抽样调查提示新疆结核病疫情依然很严重,农村疫情高于城镇,维吾尔族患病率高于其他少数民族和汉族,公众结核病防治知识知晓率尚有待提高[8]。痰培养阳性的菌株中有99例为结核分枝杆菌,采用MLVA进行基因分型及BioNumerics5.0数据库进行聚类分析。研究中发现99株菌株可分为2个基因群(Ⅰ、Ⅱ),其中,仅基因群I中有2株为同一簇,成簇率为1.5%,99株菌株中有97株呈现“唯一”基因型。通过基因聚类分析,提示新疆的结核分枝杆菌呈现明显的VNTR基因多态性,且主要流行菌株为北京基因型,但成簇率不高,说明新疆结核病的近期传播情况表现得不明显。
本研究地区结核基因成簇率偏低,考虑主要与以下几点有关:首先,新疆结核病的发生主要是由既往感染结核复发造成,存在较小比例的近期传播。是否发生结核病主要受到感染结核杆菌毒力的大小和自身抵抗力高低的影响,结核杆菌毒力强而身体抵抗力又低则容易发生结核病。人体初次受到结核杆菌感染后,通常绝大多数人没有任何症状,也不发生结核病,但在短期内(一般认为是五年)没有进展成为结核病患者的个体,在未来的日子里仍然有可能通过外源性的继发感染或者是内源性的复燃而发病。其次,本研究的99株菌株中北京基因型占66.7%,低于全国水平(85%),这可能是新疆为多民族聚集地,“古老”型北京菌株在新疆等少数民族群体中的比例显著高于汉族人群[24]。而早期的“古老”型菌株仅低频存在于俄罗斯及北美一带,后由欧亚大陆人群扩张向内陆地区迁移[25]。有研究表明“古老”型北京菌株间的遗传差异性较大,而“现代”型北京菌株间的遗传距离较小,这和新疆的结核菌株存在明显的基因多态性一致[26]。对NRAMP1基因13个SNP在高加索人群研究表明[27-28],有2个SNP与高加索人群结核病易感性相关,这在一定程度上说明了新疆的维吾尔族人群可能具有较高的易感性[29-32]。本研究尚存在较多的非北京基因型菌株,广西及浙江的研究表明其与多重耐药相关[33-34]。再次,结核病的诊断金标准为痰涂片镜检和痰结核菌培养,然而涂片敏感性只有30%,且未反复留取痰标本送检及培养,可能造成了部分阳性结核菌标本的丢失或遗漏,使纳入的标本量较少。最后,本研究中调查地区主要为南疆,生活经济及卫生服务水平相对落后,人口流动性较小,该地区患者大多为既往结核治疗不充分的结核病患者,使结核杆菌的传播力度会有所下降。新疆地域辽阔,自然环境差别较大,本研究覆盖了新疆12个地市的20个县,患者来源广,且较分散,采用随机抽样的方式,但研究获得的样本量不充分,部分患者可能已经在医院或当地卫生所接受治疗,可能导致基因成簇率低。北京家族菌株之间可以发生近期传播而成簇,这一点已在先前的研究中发现,但其传播机制还需通过进一步的实验研究及扩大的样本量加以确认。
本研究对获得的菌株进行MLVA基因分型分析,发现新疆结核分枝杆菌存在明显的VNTR基因多态性,主要流行株为北京基因型,同时还发现存在非北京基因型,本研究为今后新疆的结核菌分子生物学进一步深入研究及探讨结核的传播机制提供了方向。
1 杨春娥, 冯喜英. 结核病易感基因的研究现状[J/CD]. 中华肺部疾病杂志(电子版), 2016, 9(2): 207-209.
2 Palos C, Bispo A, Rodrigues P, et al. Electronic epidemiological query on admission: early detection of high risk patients for pulmonary tuberculosis[J]. Antimicrob Resist Infect Control, 2015, 4(Suppl 1): 61.
3 Sargazi A, Sepehri Z, Sargazi A, et al. How much Sistan was successful in tuberculosis control? [J]. Antimicrob Resist Infect Control, 2015, 4(Suppl 1): 101.
4 Sargazi A, Sepehri Z, Sagazi A, et al. Eastern Mediterranean Region tuberculosis economic burden in 2014[J]. Antimicrob Resist Infect Control, 2015, 4(Suppl 1): 102.
5 董毅, 吴利先. VNTR技术用于大理地区结核分枝杆菌基因分型的研究[J]. 大理学院学报, 2014, 13(8): 23-25.
6 Organization WH. Global Tuberculosis Control: WHO Report 2011[J]. Global Tuberculosis Control Who Report, 2011, 36(5): 497-498.
7 WHO. Global tuberculosis report 2015. WHO, 2015.
8 杨津明, 杰恩斯·斯马胡勒, 邰新蓉, 等. 新疆维吾尔自治区2010-2011年结核病流行病学抽样调查结果分析[J]. 中国防痨杂志, 2013, 35(12): 960-964.
9 Barnes PF, Cave MD. Molecular epidemiology of tuberculosis[J]. N Engl J Med, 2003, 349(12): 1149-1156.
10 Kamerbeek J, Schouls L, Kolk A, et al. Simultaneous detection and strain differentiation ofMycobacteriumtuberculosisfor diagnosis and epidemiology[J]. J Clin Microbiol, 1997, 35(4): 907-914.
11 Cowan LS, Mosher L, Diem L, et al. Variable-number tandem repeat typing ofMycobacteriumtuberculosisisolates with low copy numbers of IS6110 by using mycobacterial interspersed repetitive units[J]. J Clin Microbiol, 2002, 40(5): 1592-1602.
12 Bertrand S, De Lamine de Bex G, Wildemauwe C, et al. Multi locus variable-number tandem repeat (MLVA) typing tools improved the surveillance ofSalmonellaenteritidis: A 6 Years retrospective study[J]. PLoS One, 2015, 10(2): e0117950.
13 Pruvost O, Magne M, Boyer K, et al. A MLVA genotyping scheme for global surveillance of the citrus pathogenXanthomonascitri pv. citri suggests a Worldwide Geographical Expansion of a single genetic lineage[J]. PLoS One, 2014, 9(6): e98129.
14 Bouklata N, Supply P, Jaouhari S, et al. Molecular typing ofMycobacteriumtuberculosiscomplex by 24-Locus based MIRU-VNTR typing in conjunction with spoligotyping to assess genetic diversity of strains circulating in morocco[J]. PLoS One, 2015, 10(8): e0135695.
15 He SJ, Shi KY, Guo XZ, et al. Properties ofMethyltetrahydrophthalicanhydride-cured epoxy resin modified with MITU[J]. Polymers for Advanced Technologies, 2009, 420(2): 130-134.
16 Liu Y, Shi X, Chen Q, et al. The evaluation and application of multilocus variable number tandem repeat analysis (MLVA) for the molecular epidemiological study of Salmonella enterica subsp. enterica serovar Enteritidis infection[J]. Ann Clin Microbiol Antimicrob, 2016, 15: 4.
17 Lienemann T, Kyyhkynen A, Halkilahti J, et al. Characterization ofSalmonellaTyphimuriumisolates from domestically acquired infections in Finland by phage typing, antimicrobial susceptibility testing, PFGE and MLVA[J]. BMC Microbiol, 2015, 15: 131.
18 Brossier F, Sola C, Millot G, et al. Comparison of a semiautomated commercial repetitive-sequence-based PCR method with spoligotyping, 24-Locus mycobacterial interspersed repetitive-unit-variable-number tandem-repeat typing, and restriction fragment length polymorphism-based analysis of IS6110 forMycobacteriumtuberculosistyping[J]. J Clin Microbiol, 2014, 52(11): 4082-4086.
19 Liu Y, Tian M, Wang X, et al. Genotypic diversity analysis ofMycobacteriumtuberculosisstrains collected from Beijing in 2009, using spoligotyping and VNTR typing[J]. PLoS One, 2014, 9(9): e106787.
20 Xia E, Teo YY, Ong RT, et al. Spoligotyping and drug resistance patterns ofMycobacteriumtuberculosisisolates from five provinces of Iran[J]. Genome Med, 2016, 8(1): 19.
21 王晔茹, 徐潇, 崔生辉, 等. 多位点可变数量串联重复序列分析在细菌分子分型中的应用[J]. 中国食品卫生杂志, 2013, 25(2): 185-189.
22 Pang Hui, Tong Jin, Liu HC, et al. Molecular characterization and drug-resistance ofMycobacteriumtuberculosisstrains in Xuzhou,China[J]. Biomed Environ Sci, 2014, 27(12): 960-964.
23 焦伟伟, 李兆娜, 孙琳, 等. 多位点数目可变串联重复序列分析在北京基因型结核分枝杆菌基因分型中的应用[J]. 中华检验医学杂志, 2008, 31(11): 1249-1252.
24 Luo T, Comas I, Luo D, et al. Southern east Asian origin and coexpansion ofMycobacteriumtuberculosisBeijing family with Han Chinese[J]. Proc Natl Acad Sci USA, 2015, 112(26): 8136-8141.
25 Milan SJ, Hauge KA, Kurepina NE, et al. Expanded geographical distribution of the N family ofMycobacteriumtuberculosisstrains within the United States[J]. J Clin Microbiol, 2004, 42(3): 1064-1068.
26 Mokrousov I, Narskaya O, Otten T, et al. Phylogenetic reconstruction withinMycobacteriumtuberculosisBeijing genotype in northwestern Russia[J]. Ressearch in Microbiology, 2002, 153(10): 629-37.
27 Fernández-Mestre M, Villasmil, Takiff H, et al. NRAMP1 and VDR gene polymorphisms in susceptibility to tuberculosis in venezuelan population[J]. Dis Markers, 2015, 2015: 860628.
28 Wu F, Zhang W, Zhang L, et al. NRAMP1, VDR, HLA-DRB1, and HLA-DQB1 gene polymorphisms in susceptibility to Tuberculosis among the Chinese Kazakh population: A case-controls study[J]. Biomed Res Int, 2013, 2013: 484535.
29 Kimonis V. Clinical utility and dilemmas of SNP microarray testing[J]. Mol Cytogenet, 2014, 7(Suppl 1): 134.
30 马麦卷, 刘玮, 曹务春, 等. 结核病易感基因研究进展[J]. 中华流行病学杂志, 2011, 32(7): 650-656.
31 Chen M, Liang Y, Li W, et al. Impact of MBL and MASP-2 gene polymorphism and its interaction on susceptibility to tuberculosis[J]. BMC Infect Dis, 2015, 15: 151.
32 Etokebe GE, Bulat-Kardum L, Munthe LA, et al. Association of variable number of tandem repeats in the coding region of the FAM46A Gene, FAM46A rs11040 SNP and BAG6 rs3117582 SNP with susceptibility to tuberculosis[J]. PLoS One, 2014, 9(3): e91385.
33 王晓萌, 刘志广, 柳正卫, 等. 70株浙江省结核分枝杆菌临床分离株Spoligotyping基因分型研究[J]. 中国预防医学杂志, 2008, 9(11): 946-949.
34 刘飞鹰, 刘志广, 王喜文, 等. Spoligotyping对广西地区208株结核分枝杆菌临床分离株的基因分型[J]. 中国人兽共患病学报, 2007, 23(12): 1226-1230.
(本文编辑:黄红稷)
龚新记,李月华,姚丽丹,等. MLVA用于新疆部分地区结核分枝杆菌基因分型的初步研究[J/CD]. 中华肺部疾病杂志(电子版), 2017, 10(3): 304-308.
Preliminary study on genotype ofMycobacteriumtuberculosisin some areas of Xinjiang by the multiple locus VNTR analysis
GongXinji1,LiYuehua2,YaoLidan3,Aynur·Mokomti2,LiuNianqiang2,WangLe2,WangJing4.
1DepartmentofRespiratoryMedicine,theFirstAffiliatedHospitalofXinjiangMedicalUniversity,Urumchi830000China;2InstituteforTuberculosisControlandPrevention,CentreforDiseaseControlandPreventionofXinjiangUygurAutonomouslyRegion,Urumchi830001China;3DepartmentofCriticalCareMedicine,theFirstAffiliatedHospitalofXinjiangMedicalUniversity,Urumchi830000China;4DepartmentofGeratology,theFirstAffiliatedHospitalofXinjiangMedicalUniversity,Urumchi830000China
WangJing,Email:tlfwj@163.com
Objective To explore the multiple locus VNTR analysis (MLVA) ofMycobacteriumtuberculosis. Methods It was selected that the data of the fifth TB epidemiological sampling survey in Xinjiang Uygur Autonomously Region from 2010 to 2011, and the genotyping was carried out by the classic 24 site method. The genetic clustering was analyzed by the BioNumerics5.0 database. The original strains ofMycobacteriumtuberculosis, the annulus were dissolved and suspended bacterium in 400 μl TE, 80 ℃ 1 h inactivated, 12 000 r/min, the centrifugal 10 min, abandon supernatant, suspended bacterium in 600 μl TE again; multiple site tandem repeat sequence analysis. Results There were two genetic groups in Xinjiang: gene group Ⅰ and gene group Ⅱ, 66 strains in genetic group Ⅰ (66.7%), and 33 strains in gene strain Ⅱ (33.3%). gene growp I was Beijing genotype strains 65 different genotype from gene group of 66 strains ofMycobacteriumtuberculosisⅠ, 2 strains ofMycobacteriumtuberculosisbelong to the same cluster, clusters rate was 1.5%, It was different that the gene group of Ⅱ 33 ofMycobacteriumtuberculosisstrain of MLVA pictures, clusters rate was 0. ConclusionMycobacteriumtuberculosisstrains exist obvious gene polymorphism in Xinjiang, with the majority of Beijing genotype strains, and a certain proportion of strains are not Beijing genotype, we should strengthen the monitoring and management for major epidemic strains.
Tuberculosis;Mycobacteriumtuberculosis; Multiple locus VNTR analysis; Gene polymorphism; Xinjiang areas
10.3877/cma.j.issn.1674-6902.2017.03.013
新疆重大疾病医学重点实验室开放课题基金资助项目 (SKLIB-XJMDR-2012-2,SKLIB-XJMDR-2014-14 SKLIB-XJMDR-2016-4)
2016年新疆医科大学研究生创新创业基金项目(CXCY015)
830000 新疆医科大学第一附属医院呼吸科1、重症医学科3、老年病科4
830001 新疆维吾尔自治区疾病预防控制中心·结核病预防控制中心2
王晶,Email:tlfwj@163.com
R562
B
2016-04-30)
