固相萃取定量核磁共振波谱法测定板蓝根饮片中的表告依春
2017-07-12刘晓婷禹珊袁铭郭强胜龚灿
刘晓婷++禹珊++袁铭++郭强胜++龚灿++许旭


摘要建立了固相萃取(SPE)定量核磁共振波谱(qNMR)技术测定板蓝根饮片中有效成分表告依春含量的方法。样品用水超声提取两次,采用SPE对提取液进行富集浓缩,用qNMR测定表告依春的含量。考察了样品预处理和qNMR实验条件对测定结果的影响,选择氘代二甲基亚砜为溶剂,用基准试剂邻苯二甲酸氢钾标定的2,3,5三碘苯甲酸为内标,选择脉冲宽度P1=14.1 μs,延迟时间d1=5 s,扫描次数NS=256为qNMR定量测定表告依春的最佳实验条件。表告依春的定量峰为δ 5.365~5.399 (H7b, d,1H)。结果表明,日内测量精密度(RSD)为0.5%,日间精密度为0.8%,表告依春与三碘苯甲酸峰面积比与质量比的零截距标准曲线线性相关系数为0.9991,且斜率与理论值相符。根据响应值标准偏差和标准曲线斜率法确定此法测定表告依春的检测限(LOD)为0.05 mg/g;定量限(LOQ, S/N ≥ 150)为0.19 mg/g。包括样品提取过程的表告依春的回收率为97.4%~101.7%。采用本方法测定板蓝根饮片中的表告依春的含量为<0.19~1.26 mg/g。研究结果表明,采用SPE进行富集,扩大了qNMR的应用范围,可用于低含量复杂样品的定量分析。
关键词定量核磁共振波谱法; 固相萃取; 表告依春; 板蓝根
1引 言
定量核磁共振波谱法(qNMR)不依赖于被测物的高纯度标准品、不破坏样品,仅需样品组分有1个或1组互不干扰的特征NMR峰,依据信号峰的面积正比于产生该共振峰的质子数,即可进行定量分析,具有独特的优势\[1~4\]。近年来qNMR方法被广泛用于新药研制\[1,5,6\]、中药\[7,8\]与化学药\[9~11\]的质量控制、代谢组学\[12~14\]等研究。继美国药典(19版)、英国药典(1975年版)、日本药局方(12版)和欧洲药典(第5版)之后,中国药典从2010版开始,将此技术作为法定标准收载于通则(附录)中\[15,16\]。
板蓝根是十字花科草本植物菘蓝(Isatis Indigotica Fort)的干燥根,被载入中国药典一部,广泛用于流行性感冒、乙肝等病毒性疾病的防治,具有抗菌、抗病毒、抗内毒素、调节免疫等作用\[17,18\]。研究表明,其水提物、醇提物、總生物碱具有较强抗流感病毒作用,其中表告依春被认为是抗流感病毒的主要有效成分之一\[19\]。目前监测板蓝根中表告依春的主要方法为HPLC法\[20~23\],但这些方法都需要表告依春标准品做为对照品,成本较高。
采用qNMR方法测定中药中有效成分常存在灵敏度低和干扰强的问题。Yang等\[8\]通过加入氘代三氟乙酸,将原来互相干扰的定量峰分开,来消除干扰。禹珊等\[24\]将提取的样品旋干后溶于少量氘代试剂,通过提高浓度来改善灵敏度。固相萃取(SPE)作为一种有效的样品预处理技术,具有浓缩(富集)和样品净化功能,操作简便\[25,26\]。将SPE与qNMR结合,有助于解决灵敏度低和干扰强的问题。Pieri等\[27\]将样品提取物用C18SPE柱净化富集后,以氘代DMSO洗脱,用qNMR内标法测定了朝鲜蓟叶提取物中的莱蓟苦素,并进行方法认证(没有评价SPE回收率)。Li等\[28\]将收集的42种党参样品提取后,经C18SPE柱处理,用qNMR测定了四种生物碱,但在低浓度测定回收率时的精密度较差。Moura等\[29\]将加入内标的样品溶液用C18SPE柱净化富集后,用qNMR测定环境和生物样品中的βN甲基氨基L丙氨酸。其它应用还包括测定饮茶后人体尿液中的代谢物\[30\]、污染水中的原油和正己烷\[31\]、地下水中的污染物\[32\]等。
本研究采用SPE技术净化和富集板蓝根饮片提取物,建立SPEqNMR测定表告依春含量的方法。研究结果表明,采用SPE进行富集,扩大了qNMR的应用范围,可用于低含量复杂样品的定量分析。本方法在实际测定样品时无需对照品。
2实验部分
2.1仪器与试剂
Bruker AVANCE III 500 MHz核磁共振谱仪(德国Bruker公司);U3000高效液相色谱仪(美国ThermoFisher公司);M2p高精密天平(精度0.001 mg, 德国Startoriu公司);AB204N精密天平(精度0.1 mg, 上海世义精密仪器有限公司);PS20超声波清洗仪(东莞洁康公司);800B高速台式离心机(上海安亭科学仪器厂);DC12氮吹仪(上海安谱科学仪器有限公司)。PolySery MCX固相萃取柱(500 mg,6 mL,上海安谱实验科技股份有限公司);Kromasil高效液相色谱C18色谱柱(150 mm × 4.6 mm, 瑞典Akzo Nobel公司)。
表告依春(Epigoitrin,阿法埃莎(天津)化学有限公司);2,3,5三碘苯甲酸(上海阿拉丁生化科技股份有限公司);邻苯二甲酸氢钾(基准物质,99.95%~100.05%,上海泰坦科技股份有限公司);氘代二甲基亚砜(d6DMSO,99.9%)和氘代硫酸(99.5%)均为Cambridge Isotope Lab. Inc.产品;甲醇(HPLC级,阿达玛斯试剂有限公司);正己烷、乙酸、醋酸铵为国产分析纯;磷酸、氨水为国产优级纯。实验用水为蒸馏水。
板蓝根饮片的3个样品均购自本地中药房,药材产地分别为甘肃、江苏和安徽。其中样品3有虫蛀现象。
2.2实验方法
2.2.1样品处理将板蓝根饮片磨碎,过筛,精确称取1 g左右,加入50 mL水,称重,60℃超声提取45min,放冷后再称重,加水补充损失质量,4000 r/min离心5 min,收集上清液。残渣再加入50 mL水,重复上述提取步骤。合并两次上清液,过滤并弃去初滤液,收集50 mL续滤液。
依次用6 mL甲醇、6 mL蒸馏水、6 mL乙酸乙酸铵缓冲溶液(pH 4.7)活化PolySery MCX固相萃取柱(500 mg,6 mL)。將50 mL上述续滤液过柱,流速控制在3 s滴加1滴。用6 mL正己烷分3次淋洗柱子,每次2 mL。最后用6 mL 氨水甲醇(5∶95, V/V)溶液洗脱,洗脱液用N2吹至约0.1 mL,加入约0.4 mL氘代DMSO,再加入准确称重的2,3,5三碘苯甲酸作为内标,混匀后转移至核磁管中。
2.2.2HPLC检测条件参考2015版中国药典\[17\],选择用于考察SPE操作方法的HPLC检测条件为:Kromasil 1005C18色谱柱(150 mm × 4.6 mm),流动相为甲醇0.02% H3PO4(7∶93, V/V);流速1.0 mL/min; 进样量10 μL;柱温30℃; 检测波长245 nm。
2.2.3定量核磁共振测定核磁共振(NMR)数据采集条件:脉冲序列zg30。脉冲宽度(P1)14.1 μs,延迟时间(D1)5 s,扫描次数(NS)256次,谱宽(SW)20 ppm,中心频率(O1)3090 Hz,采样时间(AQ) 3.17 s,室温(约25℃)条件下测定NMR图谱。积分前先调平NMR基线,选择对应的峰的积分区间,每个峰积分3~5次,相对标准偏差(RSD) < 1%时取平均值。采用中国药典的绝对定量公式计算待测组分的含量Ws\[15,16\]:
Ws = Wr×(As/Ar)×(Es/Er)(1)
其中, Wr 为称取得内标物的质量, As 和Ar 分别为供试品和内标峰的峰面积, Es 和Er 分别为供试品和内标物的质子当量重量(质量)。
3结果与讨论
3.1样品制备方法的选择
3.1.1样品提取条件的优化文献报道的板蓝根药材(饮片)的提取方法主要有回流法\[33\]、煎煮法\[34\]和超声波辅助提取法\[20~23\],其中应用最广的是超声波辅助提取法。本研究采用2.2.2节的HPLC检测条件,考察了提取条件对SPE的影响。
比较了煎煮和超声提取两种方法对表告依春的提取效率。煎煮法采用中国药典2015版一部板蓝根供试品溶液的制备方法\[17\],重复提取两次。超声提取法按照2.2.1节的方法提取。用HPLC测定煎煮提取液中表告依春的峰面积,结果表明,均明显小于超声提取液的峰面积,所以后续实验选择超声提取方法。
取超声提取两次的残渣,再次超声提取,提取液中表告依春的峰面积约为第一次提取的1/70。因此,本实验选定的提取次数为2次。
选择超声波提取方法, 考察了超声时间(10~120 min)对表告依春的提取情况的影响。超声提取45 min时,表告依春峰面积较高,继续延长超声提取时间,响应值差异不大。故超声提取时间选择45 min。
3.1.2固相萃取条件的选择表告依春溶解在水中呈弱碱性。比较了MCX柱(混合型强阳离子交换柱)、C18柱两种SPE柱的效果。C18柱在上样和淋洗过程中均有大量表告依春流出,而MCX 柱对表告依春有较好保留,因此选用 MCX 固相萃取柱。
分别采用 100 mmol/L乙酸(pH 2.9)、2.5 mmol/L乙酸(pH 3.7)和乙酸乙酸铵缓冲溶液(pH 4.7)活化MCX 固相萃取柱。使用前两种溶液时,表告依春在上样时都会流出,而乙酸乙酸铵缓冲溶液能使表告依春在上样时完全保留在柱上,所以选择乙酸乙酸铵缓冲溶液活化柱子。将50 mL续滤液过柱,取第5~50 mL流出液检测,均未检测到表告依春。
考察甲酸0.1 mol/L乙酸(1∶1, V/V)、乙酸乙酸铵缓冲溶液、水、冰水、乙酸乙酯、二氯甲烷、四氢呋喃、正己烷、异丙醇和无水乙醚对MCX 柱子淋洗的影响。结果表明,使用冰水和正己烷时,表告依春的保留效果最好,其中使用正己烷时完全没有漏出。故选择使用正己烷为淋洗剂。
考察了甲醇5%氨水(1∶1, V/V)、甲醇氨水(95∶5, V/V)、氨水对表告依春的洗脱效果,最终选择甲醇氨水(95∶5, V/V)溶液为洗脱剂,用量为6 mL。
综上所述,固相萃取的最终条件为:6 mL甲醇、6 mL蒸馏水、6 mL乙酸乙酸铵缓冲溶液(pH 4.7) 活化;板蓝根续滤液50 mL上样;用6 mL正己烷淋洗;6 mL 氨水甲醇(5∶95, V/V)洗脱。
3.2氘代试剂的选择
将得到的6 mL洗脱液用氮气吹干,分别考察了氘代二甲基亚砜和氘代氯仿对所得产物的溶解性,结果表明,氘代二甲基亚砜能很好地溶解产物,而氘代氯仿并不能够完全溶解产物。所以选择氘代二甲基亚砜作为溶剂。
3.3内标物的选择
除2,3,5三碘苯甲酸外,其它常用内标物,如邻苯二甲酸氢钾、顺丁烯二酸、反丁烯二酸、对苯二甲醛、苯甲酸等的部分峰均与表告依春样品峰有重叠,因此选择2,3,5三碘苯甲酸作为内标物。
3.4信号归属及定量峰的选择
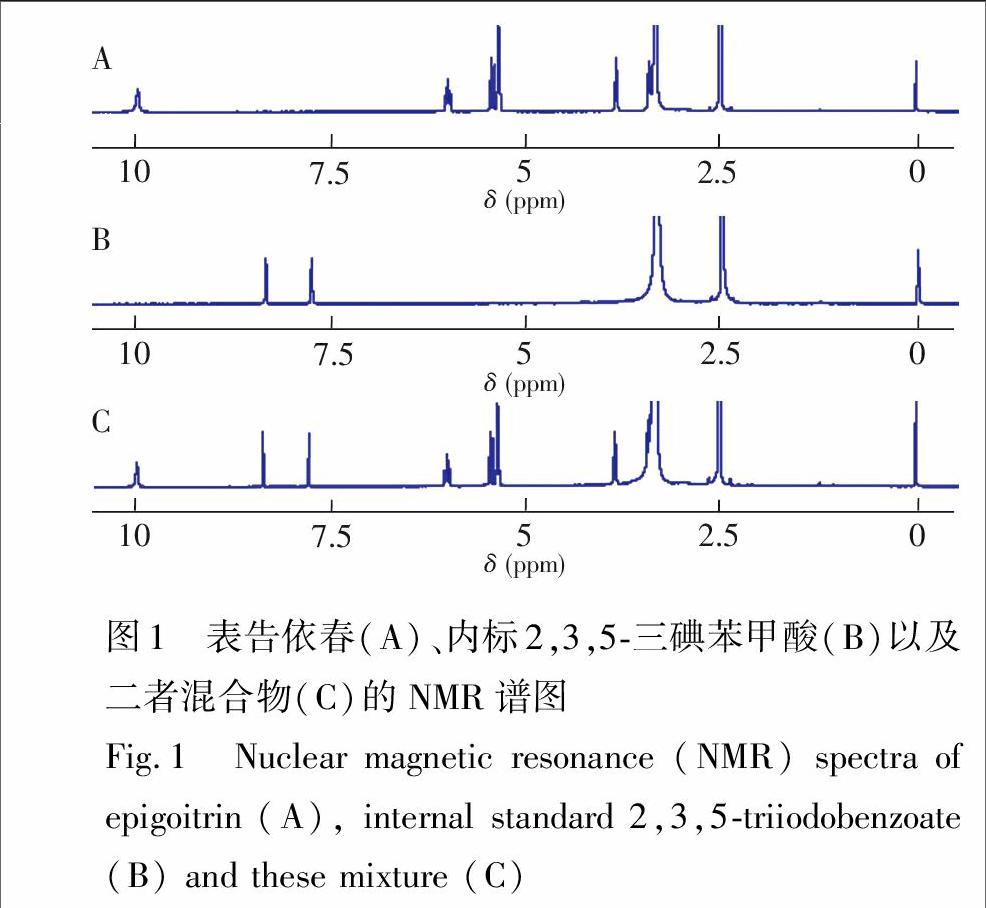
3.4.1NMR峰归属用2.2.3节的方法分别测定表告依春()、内标以及两者混合物的NMR谱图。参考文献\[35\],归属混合物(图1C)中的各信号峰, 表告依春峰: δ 3.818~3.857 (H4, t,1H)和δ 3.431~3.396 (H4′,t,1H); δ 5.310~5.324 (H5, q,1H); δ 5.343~5.356 (H7a(顺式), d,1H); δ 5.400~5.434 (H7b(反式), d,1H); δ 5.950~6.018 (H6, m,1H)。2,3,5三碘苯甲酸峰: δ 7.748~7.746 (d,1H); δ 8.333~8.335 (d,1H)。
3.4.2定量峰确认测定样品1的NMR图谱,与样品内标混合物图谱(图2C)比较。此时,内标峰位置由δ 7.748~7.746 (d,1H)和δ 8.333~8.335 (d,1H)移至δ 7.313~7.314 (d,1H)和δ 7.994 (d,1H),发生了明显变化。考虑到洗脱液为碱性,在样品中加入氘代硫酸后再测试NMR谱图,发现加入少量氘代硫酸即可以使化学位移回复到原来位置,单独采用标样也有同样现象,从而确认这种变化的原因是SPE洗脱剂含有的氨水成分,提高了NMR待测样品溶液pH值,造成NMR峰的偏移。Fan等\[36\]也用氘代盐酸调节溶液pH值,改变特征峰的化学位移。考虑样品NMR谱在δ 7.9~8.0化学位移处基线较平坦(图2B), δ 7.994(d,1H)与样品峰没有重叠,所以选择其为内标峰(图2C中信噪比较高的峰1)。在此实验条件下,NMR峰分离完全,可以满足qNMR测定要求,后续的定量实验中未用氘代硫酸调节pH值。
因此,选择内标峰δ 7.994(d,1H)对应的积分范围为δ 7.962~8.022 (圖2C中IS1),定量峰δ 5.365~5.399(d,1H)对应的积分范围为δ 5.339~5.420 (图2C中的1)。积分范围的选择方法为在对应质子峰起止处放大图谱,取峰形轮廓线与完全水平的基线刚好重合处为起止点,并固定每组峰的积分范围。
3.4.3NMR实验参数的影响 分别考察了脉冲宽度P1、延迟时间d1和扫描次数NS对积分面积的影响。当P1≥14.1 μs, d1=5 s, NS≥256时,信噪比可以达到定量要求(S/N≥150),且Ar与As趋于稳定。故选定核磁参数P1=14.1 μs, d1=5 s, NS=256。3.4.4对2,3,5三碘苯甲酸和表告依春纯品的含
量校正在2.2.2节实验条件下,以氘代DMSO为溶剂,基准物质邻苯二甲酸氢钾的NMR峰δ 7.493~7.511 (H4,5,dd,2H)与三碘苯甲酸和邻苯二甲酸氢钾纯品的NMR定量峰互不干扰。采用qNMR绝对定量模式对两种纯品进行含量校正。重复实验6次,取平均值。实验测得2,3,5三碘苯甲酸含量为99.94%,RSD为0.6%;表告依春纯品含量为97.53%,RSD为0.9%。以下验证实验中这两种化合物使用校正后的含量值。
3.5方法验证
3.5.1精密度取样品1,按照2.2.1节样品预处理方法制成待测样品,按照2.2.3节方法测得日内精密度(RSD)为0.5%(n=6),日间RSD为0.8%(n=6),表明qNMR较好的精密度以及样品良好的稳定性。
3.5.2线性与分析性能称取10组表告依春样品(0.5805~1.215 mg)和约1.5 mg内标,超声溶于0.5 mL氘代DMSO中,按2.2节方法测定NMR谱图。以表告依春与三碘苯甲酸质量比为横坐标(x),两者的峰面积比为纵坐标(y),线性拟合。零截距标准曲线:y=3.7785x, r=0.9991 (不过原点的拟合方程为: y=3.7734x+0.0036, r=0.9991。根据公式(1)计算表告依春线性曲线的理论斜率(Es/Er)为3.8691,而实验中得到的斜率与其接近,说明可以采用绝对定量公式(1)计算表告依春的含量。
3.5.3检出限和定量限根据2015版中国药典四部通则\[15\],qNMR的检出限LOD和定量限LOQ由基于响应值标准偏差和标准曲线斜率法的公式LOD= 3.3 σ/S,(LOQ)= 10σ/S 得到,式中σ在本实验采用标准曲线的剩余标准偏差计算。表告依春的LOD=0.029 mg, LOQ=0.088 mg。根据实验中内标的用量1.5 mg以及溶剂0.5 mL,计算得qNMR测定表告依春的LOD=0.09 g/L,LOQ=0.26 g/L。考虑到信噪比对qNMR的重复性影响较大,英国药典\[37\]对核磁定量方法要求信噪比S/N≥150,文献\[28\]中较低信噪比时重复实验的精密度RSD>2%,因此将其作为qNMR确定LOQ的指标是合理的,以样品1的NMR谱峰信噪比,根据公式(1)计算得表告依春的LOQ=0.37 g/L。取两者均满足的较大值,可以认为本方法qNMR对表告依春的LOQ=0.37 g/L,LOD=0.09 g/L。按照本实验中约0.5 mL的溶剂量和约1 g板蓝根的样品量,可以算出本SPEqNMR法测定板蓝根中表告依春含量的LOD=0.05 mg/g;LOQ=0.19 mg/g,相当于0.019% (w/w)。因此,通过SPE浓缩和富集分析物,显著提高了qNMR检测的灵敏度。
3.5.4表告依春的回收率分别准确称取4份各约1 g已知表告依春含量的样品,在每份样品中均加入准确称定的表告依春,用2.2节的方法提取并测定含量,计算回收率,结果见表1,表告依春回收率为97.42%~101.70% ,RSD=2.0% (n=4)。
3.6样品分析
实际样品的测定结果见表2。样品1和样品2符合2015版中国药典规定板蓝根饮片中的表告依春的含量测定不低于0.02%的要求\[17\],样品3可能是虫蛀的影响其含量偏低,不能满足药典的要求。
4结论
建立了固相萃取qNMR测定板蓝根饮片中的表告依春含量的方法,本方法精密度好,线性评价表明,可以直接采用绝对定量公式计算含量,定量分析结果准确。本方法将SPE与qNMR结合,通过净化样品和浓缩待测组分,提高了qNMR的定量分析灵敏度,有利于扩展qNMR的应用范围。
References
1YU XiaoBo, SHEN WenBin, XIANG BingRen. Pharm. Sci., 2010, 34(1): 17-23
于小波, 沈文斌, 相秉仁. 药学进展, 2010, 34(1): 17-23
2Pauli G F, Gdecke T, Jaki B U, Lankin D C. J. Nat. Prod., 2012, 75(4): 834-851
3Simmler C, Napolitano J G, McAlpine J B, Chen S N, Pauli G F. Curr. Opin. Biotechnol., 2014, 25: 51-59
4LIU Ying, HU ChangQin. Chinese Journal of Pharmaceutical Analysis, 2001, 21 (6): 447-452
刘 英, 胡昌勤. 药物分析杂志, 2001, 21 (6): 447-452
5Mahajan S, Singh I P. Magn. Reson. Chem., 2013, 51(2): 76-81
6Liu X , Kolpak M X, Wu J, Leo G C. Anal. Chem., 2012, 84(15): 6914-6918
7Liu N Q, Choi Y H, Verpoorte R, Kooy F. Phytochem. Anal., 2010. 21(5): 451-456
8Yang M, Wang J, Kong L. J. Pharm. Biomed. Anal., 2012. 70: 87-93
9Li Z Y, Welbeck E, Wang R F, Liu Q, Yang Y B, Chou G X, Bi K S, Wang Z T. Phytochem. Anal., 2015, 26(1): 8-14
10Staneva J, Denkova P, Todorova M, Evstatieva L. Pharm. Biomed. Anal., 2011, 54(1): 94-99
11Chauthe S K, Sharmal R J, Aqil F, Gupta R C, Singh I P. Phytochem. Anal., 2012, 23(6): 689-696
12Barding Jr G A, Salditos R, Larive C K. Anal. Bioanal. Chem., 2012, 404(4): 1165-1179
13Salem A A, Abdou I M, Saleh H A. J. AOAC Int., 2012, 95(6): 1644-1651
14Moazzami A A, Andersson R E, KamalEldin A. J. Nutr., 2007, 137(4): 940-944
15National Pharmacopoeia Committee. The Pharmacopoeia of the People′s Republic of China ( Part IV, 2015 Ed), Beijing: China Medical Science Press, 2015: 52-54, 376
國家药典委员会编. 中华人民共和国药典(四部, 2015年版). 北京: 中国医药科技出版社, 2015: 52-54, 376
16National Pharmacopoeia Committee. The Pharmacopoeia of the People′s Republic of China ( PartII, 2010 Ed), Beijing: China Medical Science Press, 2010: Appendix Page 81-83
国家药典委员会. 中华人民共和国药典(二部, 2010年版). 北京: 中国医药科技出版社, 2010: 附录81-83
17National Pharmacopoeia Committee. The Pharmacopoeia of the People′s Republic of China ( PartI, 2015 Ed), Beijing: China Medical Science Press, 2015: 205-206
国家药典委员会. 中华人民共和国药典(一部, 2015年版). 北京: 中国医药科技出版社, 2015: 205-206
18HUANG HeFei, LIU HongBing, WANG YanYan, ZHOU Gang, ZHANG Hua, HUANG ZhiXiong. Journal of Hunan Normal University(Medical Science), 2014, 11(2): 46-48
黄鹤飞, 刘红兵, 王燕燕, 周 刚, 张 烨, 黄志雄. 湖南师范大学学报(医学版), 2014, 11(2): 46-48
19XU LiHua, HUANG Fang, CHEN Ting, WU Jie. Chinese Journal of Natural Medicines , 2005, 3(6): 359-360
徐丽华, 黄 芳, 陈 婷, 吴 洁. 中国天然药物, 2005, 3(6): 359-360
20WANG Rui, YANG HaiYing, YANG QiWei, HUANG ShanJun, WANG ZhengTao. Chinese Traditional and Herbal Drugs, 2010, 41(3): 478-480
王 瑞, 杨海英, 杨琪伟, 黄山君, 王峥涛. 中草药, 2010, 41(3): 478-480
21ZHANG Ye, ZHANG Lei, WANG Yu. Chinese Journal of Pharmaceutical Analysis, 2008, 28(11): 1848-1850
张 叶, 张 蕾, 王 玉. 药物分析杂志, 2008, 28(11): 1848-1850
22AN YiQiang, JIA XiaoBin, YUAN HaiJian, SUN E, XU ZhenZhen. China Journal of Chinese Materia Medica, 2008, 33(18): 2074-2076
安益强, 贾晓斌, 袁海建, 孙 娥 , 许珍珍. 中國中药杂志, 2008, 33(18): 2074-2076
23NIE LiXing, WANG GangLi, DAI Zhong, LIN RuiChao. Chinese Journal of Chromatography, 2010, 28(10): 1001-1004
聂黎行, 王钢力, 戴 忠, 林瑞超. 色谱, 2010, 28(10): 1001-1004
24 YU Shan, GUO QiangSheng, WANG HuiLin, GAO JianPing, XU Xu. Chinese Journal of Analytical Chemistry, 2015, 43(1): 69-74
禹 珊, 郭强胜, 王会琳, 高建平, 许 旭. 分析化学, 2015, 43(1): 69-74
25AndradeEiroa A, Canle M, LeroyCancellieri V, Cerdà V. TRACTrends Anal. Chem., 2016, 80: 655-667
26HE JiuMing, DAI DongMei, LIU Ying, SUN RuiXiang, ABLIZ Zeper. Chinese J. Anal. Chem., 2009, 37(suppl): D185
贺玖明, 代冬梅, 刘 影, 孙瑞祥, 再帕尔·阿不力孜. 分析化学, 2009, 37(增刊): D185
27Pieri V, Stuppner H. Planta Med., 2011, 77(15): 1756-1758
28Li C Y, Xu H X, Han Q B, Wu T S. J. Chromatogr. A, 2009, 1216(11): 2124-2129
29Moura S, Ultramari M D A, Paula D M L D, Yonamine M, Pinto E. Toxicon, 2009, 53(5): 578-583
30 van der Hooft J J J, de Vos R C H, Mihaleva V, Bino R J, Ridder L, de Roo N, Jacobs D M, van Duynhoven J P M, Vervoort J. Anal. Chem., 2012, 84(16): 7263-7271
31Wagner L, Kall C, Fridjonsson E O, May E F, Stanwix P L, Graham B F, Carroll M R J, Johns M L. Meas. Sci. Technol., 2016, 27(10): 105501
32Godejohann M, Heintz L, Daolio C, Berset J D, Muff D. Environ. Sci. Technol., 2009, 43(18): 7055-7061
33MA Li, SUN Qin, LI You, XIAO XiaoHe. Chinese Journal of Pharmaceutical Analysis, 2010, 30(9): 1642-1645
马 莉, 孙 琴, 李 友, 肖小河. 药物分析杂志, 2010, 30(9): 1642-1645
34WANG XiaoLiang, CHEN MingHua, WANG Fang, BU PengBin, LIN Sheng, ZHU ChengGen, LI YuHuan, JIANG JianDong, SHI JianGong. China Journal of Chinese Materia Medica, 2013, 38(8): 1172-1182
王晓良, 陈明华, 王 芳, 卜鹏滨, 林 生, 朱承根, 李玉环, 蒋建东, 石建功. 中国中药杂志, 2013, 38(8): 1172-1182
35 Gardrat C, Latxague L, Picard J P. J. Heterocyclic Chem., 1990, 27(3): 811-812
36 Fan G, Zhang M Y, Zhou X D, Lai X R, Yue Q H, Tang C, Luo W Z, Zhang Y. Analytica Chimica Acta, 2012, 747: 76-83
37The British Pharmacopoeia Commission. British Pharmacopoeia (2013 Ed). London: The Stationary Office, 2013: Appendix IIC Nuclear Magnetic Resonance Spectrometry
