聚谷氨酸修饰电极测定食品中的麦芽酚
2017-06-23邓振旭
朱 琪,邓振旭
(1.菏泽学院化学化工系,山东菏泽 274015;2.菏泽学院生命科学系,山东菏泽 274015)
聚谷氨酸修饰电极测定食品中的麦芽酚
朱 琪1,邓振旭2,*
(1.菏泽学院化学化工系,山东菏泽 274015;2.菏泽学院生命科学系,山东菏泽 274015)
建立一种用聚谷氨酸修饰电极测定麦芽酚的新方法。制备了聚谷氨酸修饰电极,用循环伏安法研究了麦芽酚在聚谷氨酸修饰电极上的电化学行为。实验表明:聚谷氨酸修饰电极对麦芽酚的电化学氧化具有明显催化作用,与未修饰的玻碳电极相比,麦芽酚在聚谷氨酸修饰电极上的氧化峰电流明显增大;在pH8.0的磷酸氢二钠-柠檬酸缓冲溶液(sodium hydrogen phosphate-citric acid buffer solution,PBS)中,麦芽酚在聚谷氨酸修饰电极上呈现不可逆的氧化峰,氧化峰电流与麦芽酚浓度成正比,线性范围为2.40×10-6~6.61×10-4mol/L,检出限为8.0×10-7mol/L。用该种方法测定了面包、饮料、啤酒中的麦芽酚,回收率在98.8%~103.7%之间,结果满意。该方法灵敏、准确、实用,对食品中麦芽酚的测定具有实际意义和应用前景。
聚谷氨酸修饰电极,麦芽酚,循环伏安法
麦芽酚,化学名称:3-羟基-2-甲基-4-吡喃酮,是一种广谱香味增效剂,有很强的增甜增鲜、增香、固香和抑制苦、酸、涩味的作用,作为食品增香剂常添加于焙烤食物,冰淇淋和糖果等[1]。但是一些资料显示麦芽酚具有诱导细胞死亡的毒性作用[2],大剂量的摄入会导致恶心头痛、呕吐、呼吸困难等症状,甚至可能伤及肝肾,还有可能有弱的致突变活性,对人体健康危害较大。世界卫生组织食品添加剂专业委员会和联合国粮食与农业组织议定,以体重计,人均每天麦芽酚摄入量不能超过2 mg/kg[3]。所以寻找能够快速、准确、方便的检测食品中麦芽酚含量的方法是十分必要的。目前,麦芽酚测定方法主要有气相色谱-质谱法[4]、HPLC-安培检测法[5]、紫外-可见光度法[6]、FIA-电化学发光法[7]等。而使用电化学分析方法操作比较简便,灵敏度高,分析的速度也较快[8-9],但目前已报道的电化学修饰电极对麦芽酚的测定方法还很少。
本工作研究了测定麦芽酚的电化学新方法,制备了聚谷氨酸修饰电极,研究了麦芽酚在聚谷氨酸修饰电极上的电化学行为,建立了测定麦芽酚的新方法,该方法操作简单,并且测定线性范围较宽,检出限较低。该方法灵敏、准确、实用,对食品中麦芽酚的测定具有实际意义和应用前景。
1 材料与方法
1.1 材料与仪器
CH1660C电化学工作站 上海辰华仪器有限公司;KH-100DB型超声波清洗器 昆山市超声仪器有限公司;实验用聚谷氨酸修饰电极做工作电极,对电极用铂丝电极,Ag/AgCl电极为参比电极。
麦芽酚(分析纯) 上海市阿拉丁试剂有限公司,配制成浓度为8.00×10-3mol/L的溶液;谷氨酸(分析纯) 上海新兴医药保健品科技开发中心,配制成浓度为1.00×10-2mol/L的溶液;PBS缓冲溶液 由0.2 mol/L磷酸氢二钠(分析纯)与0.1 mol/L柠檬酸(分析纯)(Na2HPO4-C6H8O7·H2O)配制成;溶液均用二次去离子水配制。
1.2 样品制备
取10.0 g面包碾碎,加20.0 mL二次蒸馏水浸泡半个小时,抽滤,滤渣重复操作两次,合并滤液,定容到50 mL,制成样品溶液,5 ℃保存备用。
取某品牌格瓦斯饮料100 mL,加热微沸5 min,驱除二氧化碳。定容至100 mL制成样品溶液,5 ℃保存备用。
取某品牌啤酒100 mL,加热微沸5 min,驱除二氧化碳。定容至100 mL制成样品溶液,5 ℃保存备用。
1.3 制备修饰电极
首先在湿润的粒度为1000的金相砂纸上将Φ=3.8 mm玻碳电极磨光,然后用粒径为0.05 μm的Al2O3悬乳液抛光,抛光后的玻碳电极呈镜面效果即可,用亚沸蒸馏水冲洗干净,依次用1∶1(v/v)HNO3、无水乙醇、亚沸蒸馏水超声清洗。将处理后的电极放入含有0.5×10-3mol/L谷氨酸的pH6.0 PBS溶液中,以玻碳电极为工作电极,铂丝电极为对比电极,Ag/AgCl电极为参比电极,在-0.1~2.0 V范围内循环扫描13圈,扫描速度为100 mV/s,取出,用亚沸蒸馏水冲洗干净。
1.4 实验方法
1.4.1 最佳聚合条件的选择 以玻碳电极为工作电极,铂丝电极为对比电极,Ag/AgCl电极为参比电极,在含有一定浓度谷氨酸的PBS底液中用循环伏安法进行聚合。用得到的修饰电极为工作电极,继续采用三电极体系,在含有1 mL 8.00×10-3mol/L麦芽酚的PBS(pH5.0)溶液中,在0.3~1.2 V的电位范围内,以100 mV/s的扫描速度进行扫描,记录循环伏安图。分别依次改变聚合底液的pH、聚合周数、起始电位、终止电位及扫描速率进行电极修饰,根据所测麦芽酚氧化峰电流的大小,选择修饰电极最佳制备条件。
1.4.2 测定麦芽酚的条件选择 用在最佳聚合条件下制备的聚谷氨酸修饰电极为工作电极,铂丝电极为对比电极,Ag/AgCl电极为参比电极,分别依次改变测试底液的pH、高低电位、扫描速度、搅拌时间进行扫描,记录循环伏安图。
测定麦芽酚底液pH的选择:固定其它条件不变,分别用不同pH的PBS为底液,加入1 mL 8.00×10-3mol/L麦芽酚,以聚谷氨酸修饰电极为工作电极,铂丝电极为对比电极,Ag/AgCl电极为参比电极,在0.3~1.2 V的电位范围内,以100 mV/s的扫描速度进行扫描,记录循环伏安图。
其它测定麦芽酚的条件选择方法与选择pH的方法相似,固定其它条件不变,只改变待选择条件进行实验。
1.4.3 回收率测定 取5.0 mL面包样品溶液,加入2 mL EDTA,2 mL NH4F,掩蔽干扰离子,加入10 mL pH8.0的PBS缓冲溶液和1 mL的水,进行加标回收实验。
取1.0 mL格瓦斯样品溶液,加入2 mL EDTA,2 mL NH4F,掩蔽干扰离子,加入10 mL pH8.0的PBS缓冲溶液和5 mL的水,进行加标回收实验。
取1.0 mL啤酒样品溶液,加入4 mL NH4F,掩蔽干扰离子,加入10 mL pH8.0的PBS缓冲溶液和5 mL的水,进行加标回收实验。
2 结果与讨论
2.1 在玻碳电极上修饰聚谷氨酸的最佳条件
选择最佳的聚合条件,分别依次改变聚合底液pH、聚合周数、起始电位、终止电位及扫描速率进行电极修饰。图1表明聚合的条件不同,修饰电极对麦芽酚的响应电流也不同。
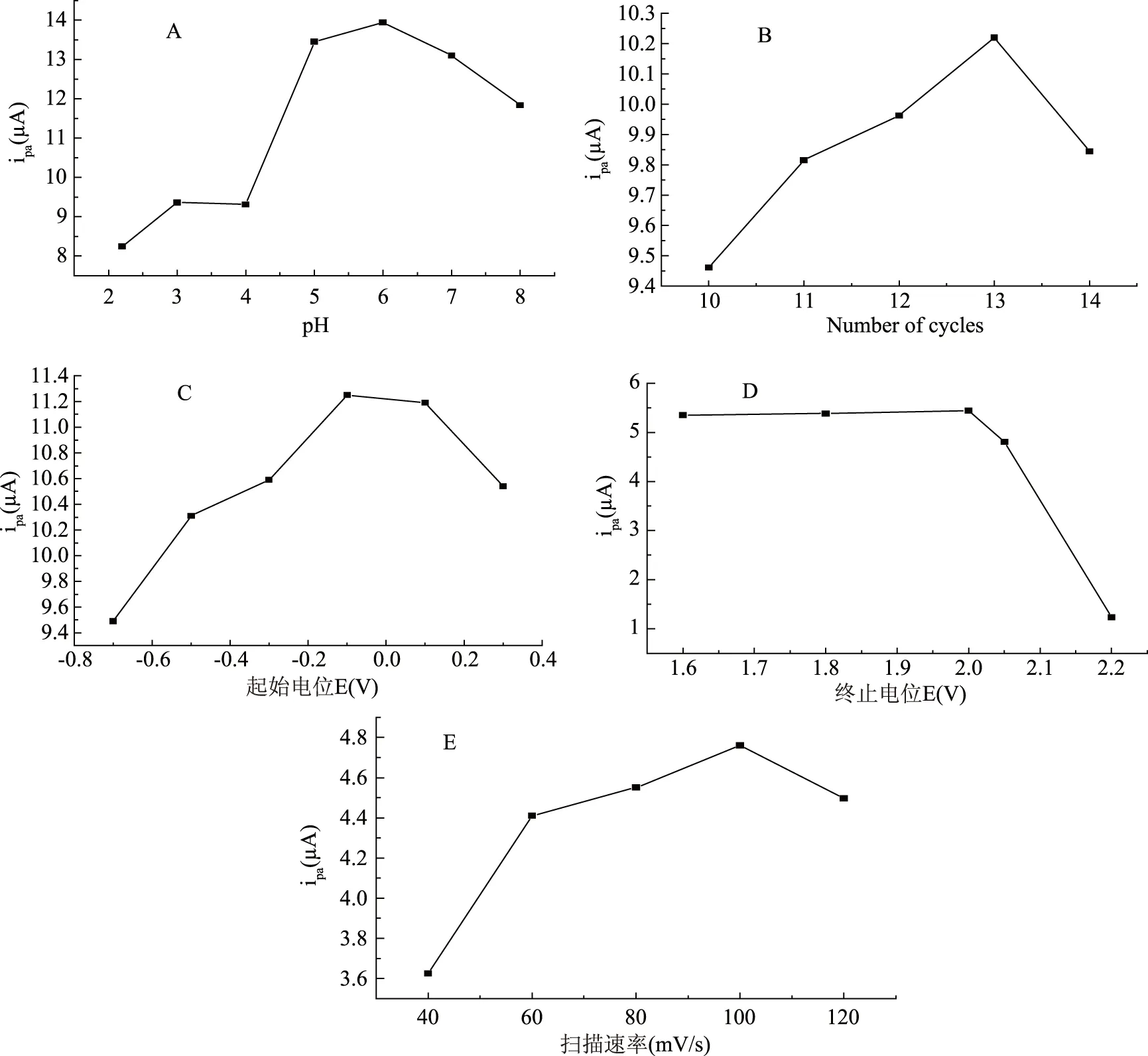
图1 在不同条件下的聚谷氨酸修饰电极上麦芽酚的氧化峰电流变化Fig.1 Variations in oxidation peak response of maltol in the poly process of glutamic acid on the graphene modified electrode
结果表明,在pH6.0的磷酸氢二钠-柠檬酸缓冲溶液中,在-0.1~2.0 V电位范围内,扫描速度为100 mV/s时,聚合13周,谷氨酸浓度为5.0×10-4mol/L时所聚合的聚谷氨酸膜对麦芽酚的测定效果最佳,氧化峰电流响应值最大,聚合图见图2。
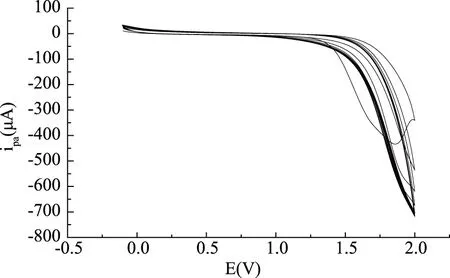
图2 制备聚谷氨酸修饰电极的循环伏安曲线Fig.2 Cyclic voltammetry curves for the preparation of poly glutamic acid modified electrode
2.2 麦芽酚在聚谷氨酸修饰电极上的电化学行为
图3为麦芽酚在裸电极(a)和聚谷氨酸修饰电极(b)上的循环伏安曲线。结果表明,麦芽酚在玻碳电极上氧化峰电流与电位分别为:ipa=-7.43 μA,Epa=0.833 V,而在聚谷氨酸修饰电极上,ipa=-10.42 μA,Epa=0.791 V,与裸电极相比麦芽酚在聚谷氨酸修饰电极的氧化峰明显增大,说明修饰电极对麦芽酚的电化学氧化具有很好的电催化作用。循环伏安曲线上无还原峰,说明在聚谷氨酸修饰电极上麦芽酚的氧化反应为不可逆过程。
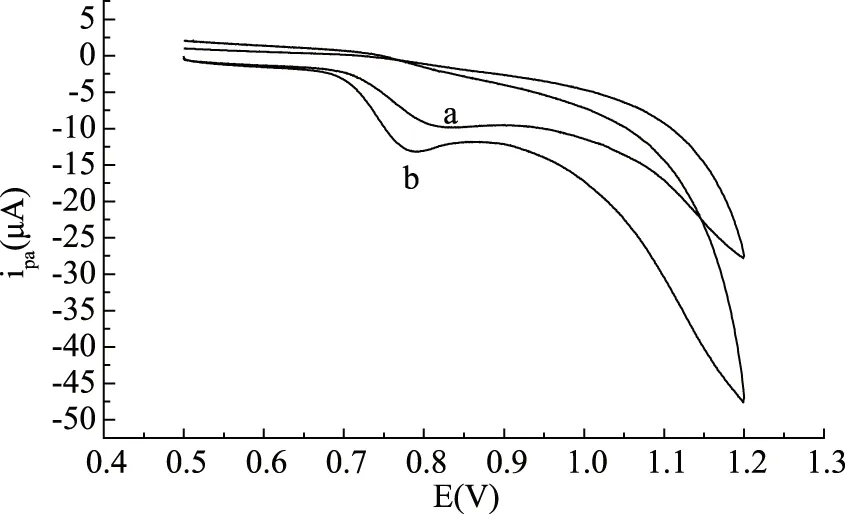
图3 麦芽酚在电极上循环伏安曲线Fig.3 Cyclic voltammograms of maltol on electrode 注:(a)裸玻碳电极;(b)修饰电极。
2.3 测定麦芽酚的条件选择
2.3.1 试液pH的影响 分别用不同pH的PBS为底液,对一定浓度的麦芽酚溶液用聚谷氨酸修饰电极进行实验。结果显示麦芽酚氧化峰电流随着溶液pH改变而变化,pH增大,氧化峰电流减小,变化过程中峰电位负移,在pH2.2时,氧化峰电流值最大(图4)。
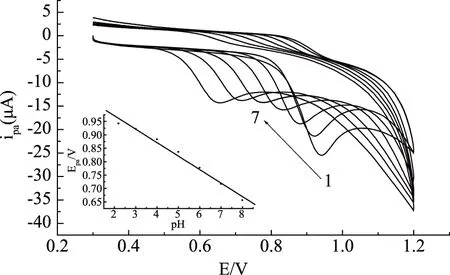
图4 麦芽酚在不同pH PBS中的循环伏安曲线Fig.4 Cyclic voltammograms of maltol in PBS of various pH注:从1到7的pH为:2.2、3.0、4.0、5.0、6.0、7.0、8.0; 内插图为氧化峰电位与测定底液pH的关系曲线,麦芽酚浓度4.0×10-4 mol/L。
在pH为2.2~8.0之间,Epa与pH呈线性关系,线性方程为:Epa=1.07-0.050pH,r=0.9927(见图4内插图)。
在对实际样品的分析测定中,由于选择EDTA作为掩蔽剂,在pH2.2的磷酸氢二钠-柠檬酸缓冲溶液中的掩蔽效果较差,影响对麦芽酚的测定。对样品在不同pH的PBS中进行实验,测定结果表明,在pH8.0的PBS中,EDTA的掩蔽效果最好,对麦芽酚浓度的测定最为准确,所以选择pH8.0的PBS作为测定底液。
2.3.2 高低电位的影响 选择测定条件时,高低电位的影响也很重要。首先,选择1.0 V为高电位,改变低电位进行实验,低电位为-0.1 V时,麦芽粉在聚谷氨酸修饰电极上的电流响应值最大。设定-0.1 V为低电位,改变高电位进行实验,当高电位为1.2 V时,麦芽粉在聚谷氨酸修饰电极上的电流响应值最大。所以,选择在-0.1~1.2 V的电位范围内进行测定。
2.3.3 扫描速度与搅拌时间的影响 以pH8.0的PBS为测定底液,固定高低电位,改变扫描速度进行实验。结果表明随着扫描速度的增大,氧化峰电位发生正移,麦芽酚的氧化峰电流也逐渐增大(图5)。氧化峰电流与扫描速率呈线性关系,当扫速在60~300 mV/s之间时,线性方程为:ipa(A)=-1.09×10-5-5.66×10-6v(mV/s),r=0.9938,说明麦芽酚在聚谷氨酸修饰电极上的电极过程为吸附过程。扫描速度为100 mV/s时,麦芽酚氧化峰峰形好,且灵敏度高,因此选择扫描速度为100 mV/s进行实验测定。
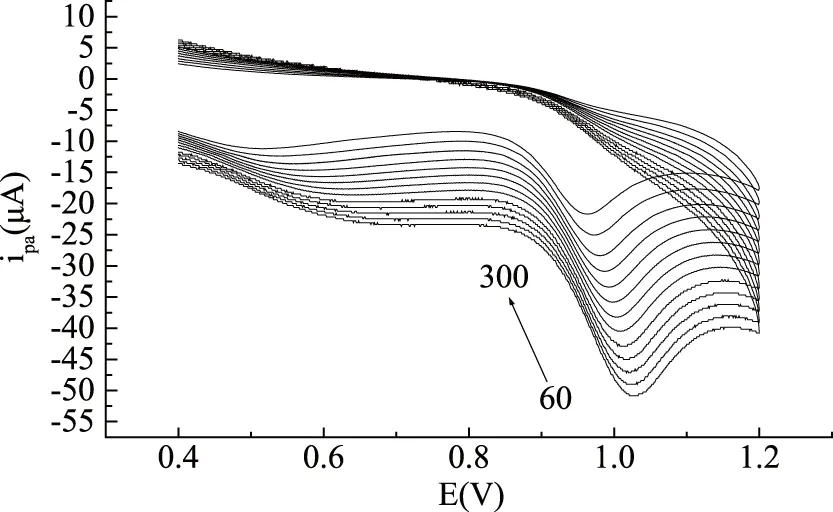
图5 麦芽酚在聚谷氨酸修饰电极上随扫速变化(60~300 mV/s)的循环伏安图Fig.5 Cyclic voltammograms of maltol on the modified electrode under different sweep rates(60~300 mV/s)
不同的搅拌富集时间对麦芽酚测定的结果影响。每20 s记录扫描循环伏安图,搅拌富集时间从20 s逐渐增大至100 s,结果发现随时间的增加,氧化峰电流逐渐减弱(图6),因此选择搅拌富集时间为20 s。
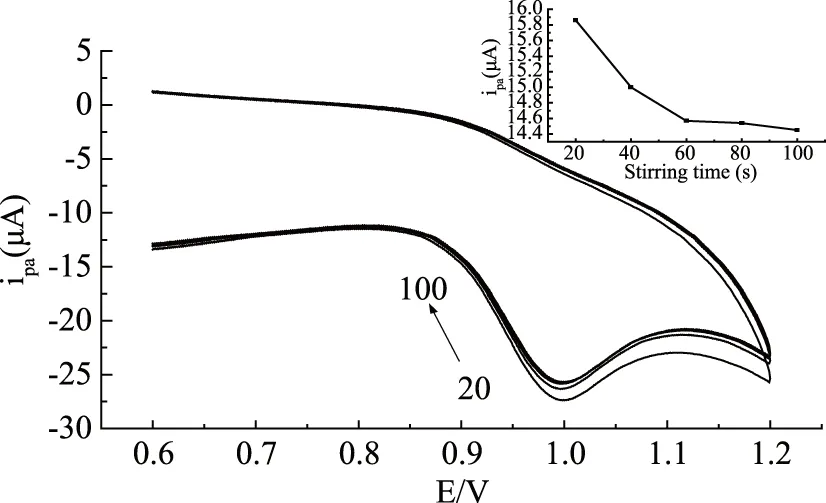
图6 麦芽酚在聚谷氨酸修饰电极上随搅拌时间变化(20~100 s)的循环伏安图Fig.6 Cyclic voltammograms of maltol on the modified electrode under different stirring time(20~100 s)
2.4 标准曲线与检出限
在pH8.0的PBS中,采用循环伏安法对麦芽酚标准溶液系列进行测定,结果表明,在2.40×10-6~2.40×10-5mol/L及2.40×10-5~6.61×10-4mol/L范围内,麦芽酚在聚谷氨酸修饰电极上的氧化峰电流与其浓度呈良好线性关系(图7),其线性方程分别为:ipa(μA)=0.08c+1.70×10-6,ipa(μA)=0.02c+3.96×10-6;相关系数分别为0.9951及0.9995,检出限为8.0×10-7mol/L。图7内插图为麦芽酚在聚谷氨酸修饰电极上不同浓度下的循环伏安曲线。
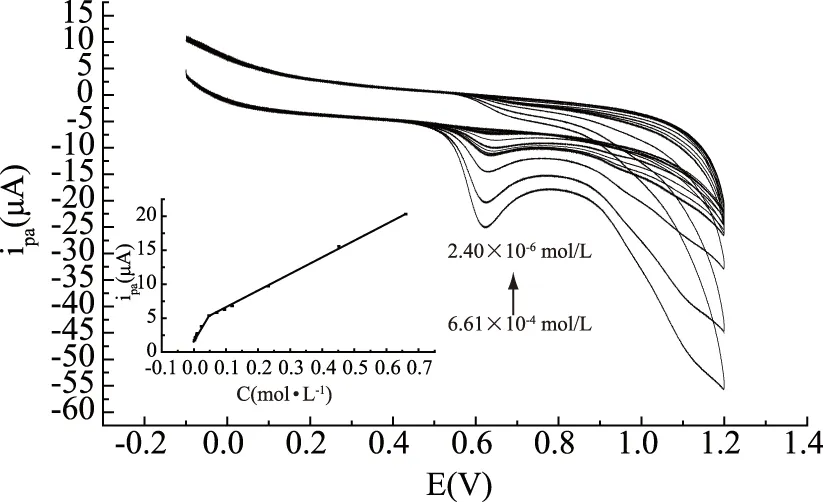
图7 不同浓度下麦芽酚在聚谷氨酸修饰电极上的循环伏安曲线Fig.7 Cyclic voltammograms of maltol at various concentrations at poly glutamic acid modified electrode注:浓度为2.40×10-6、4.80×10-6、7.20×10-6、9.60×10-6、1.20×10-5、2.40×10-5、4.80×10-5、7.20×10-5、9.60×10-5、1.20×10-4、2.33×10-4、4.53×10-4、6.61×10-4 mol/L,内插图为麦芽酚的浓度与氧化峰响应电流的关系曲线。
2.5 干扰实验
实验研究了一些可能共存物质对8.0×10-5mol/L麦芽酚测定的影响。在最佳实验条件下对8.0×10-5mol/L麦芽酚进行测定,记录循环伏安曲线;然后将100倍的Na+、K+、Zn2+、Fe3+、Ca2+、Mg2+,谷氨酸、葡萄糖、蔗糖高浓度小体积加入麦芽酚溶液中,所得循环伏安曲线与没有加入这些物质时循环伏安曲线基本重合,即电流大小不变,因此确定不干扰测定。
2.6 实际样品分析
如表1所示,采用此种方法对食品样品面包、格瓦斯饮料、啤酒中的麦芽酚含量进行测定,回收率在98.8%~103.7%之间,结果令人满意。
乙基麦芽酚是麦芽酚的同系物,结构近似,性质近似,在聚谷氨酸修饰电极上的电化学反应相同,也可以用此方法进行测定,不过二者共存时只能测定其总量。
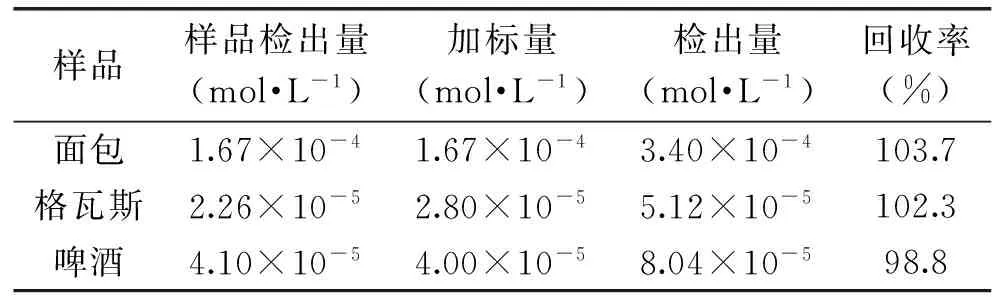
表1 样品分析结果Table 1 Analyzing result of sample
3 结论
本文制备了聚谷氨酸修饰电极,研究了麦芽酚在聚谷氨酸修饰电极上的电化学行为,结果表明聚谷氨酸修饰电极对麦芽酚的电化学反应具有显著催化作用,并且测定线性范围较宽,检出限较低。将该方法用于面包、饮料、啤酒中麦芽酚的测定,回收率较好。与其它方法比较,操作简单,成本较低,具有实际应用前景。
[1]杨海燕.多功能增香剂-麦芽酚和乙基麦芽酚[J]. 配料,1998,5(11):29-31.
[2]Hironishi M,Kordek R,Yanagihara R,et al. Maltol(3-hydroxy-2-methyl-4-pyrone)toxicity in neur-oblastoma cell linesand primary murine fetal hippocampal neuronal cultures[J].Neurodegene-ration,1996,5(4):325-329.
[3]Freydberg N,Gortner W A. The food additives book[M]. New York:Bantann Books,1982,11(2):23-25.
[4]Adahchour M,Vreuls R J J,Heijden A,et al. Trace-level determination of polar flavour compounds in butter by solid-phase extraction and gas chromatography-mass spectrometry[J]. J Chromatogr A,1999,884(1-2):295-305.
[5]Portela M J,Balugera Z G,Goicolea M A,et al. Electrochemical study of the flavour enhancer maltol.Determination in foods by liquid chromatography with amperometric detection[J]. Anal Chim Acta,1996,327(1):65-71.
[6]Kihara K. An Improved method of maltol determination for the general application and maltol distribution in plants[J].Nippon Shoyu Kenkyusho Zasshi,19982,8(2):75-79.
[7]Alonso M C S,Zamora L L,Calatayud J M. Petermination of the flavor enhancer maltol through a FIA-direct chemiluminescence procedure[J].Anal,chim. Acta,2001,438(1):157-163.
[8]周君,狄俊伟,吴莹,等.基于多壁碳纳米管化学修饰电极直接测定饮料中的麦芽[J].应用化学,2008,25(1):81-84.
[9]Du Meiju,Han Xiaogang,Zhou Zhihao,et al.Determination of Sudan I in hot chili powder by using an activated glassy carbon electrode[J]. Food Chemistry,2007,105(2):883-888.
Determination of maltol in food by cyclic voltammetry with the poly glutamic acid modified electrode
ZHU Qi1,DENG Zhen-xu2,*
(1.Department of Chemistry & Chemical Engineering,Heze University,Heze 274015,China; 2.Department of Life Sciences,Heze University,Heze 274015,China)
A novel voltammetry methodology was developed for determination of maltol using a poly glutamic acid modified electrode. The poly glutamic acid modified electrode was prepared by cyclic voltammetry. The electrochemical behavior of maltol on the poly glutamic acid modified electrode was studied. The experimental results showed that the poly glutamic acid modified electrode exhibited significant catalytic activity for the electro-oxidation of maltol,compared with the unmodified glassy carbon electrode,the oxidation peak current increased significantly at the poly glutamic acid modified electrode. In the sodium hydrogen phosphate-citric acid buffer solution(PBS)of pH8.0,an irreversible oxidation peak of matol was obtained by cyclic voltammetry,the oxidation peak current was proportional to the concentration of maltol in the range of 2.40×10-6~6.61×10-4mol/L and the detection limit was 8.0×10-7mol/L. The method was applied for the determination of maltol in food with satisfactory recoveries of 98.8%~103.7%. This method is sensitive,accurate and practical and it has practical significance and application prospects for the determination of matol in a series of food.
poly glutamic acid modified electrode;maltol;cyclic voltammetry
2016-12-08
朱琪(1987-),女,硕士研究生,助理实验师,研究方向:电化学分析,E-mail:263867584@qq.com。
*通讯作者:邓振旭(1964-),男,学士,教授,研究方向:生物电化学传感器,E-mail:dengzhenxu@126.com。
山东省自然科学基金(ZR2014BL020);山东省高校科技计划项目(J14LC55)。
TS207.3
A
1002-0306(2017)11-0305-05
10.13386/j.issn1002-0306.2017.11.050
