2型糖尿病患者肠道菌群变化及意义
2017-06-19马苏娴张锐锐王苏邓家良杨禄袁国跃
马苏娴,张锐锐,王苏,邓家良,杨禄,袁国跃
(1江苏大学附属医院,江苏镇江212001;2天益健康科学研究院)
2型糖尿病患者肠道菌群变化及意义
马苏娴1,张锐锐2,王苏1,邓家良1,杨禄2,袁国跃1
(1江苏大学附属医院,江苏镇江212001;2天益健康科学研究院)
目的 探讨2型糖尿病(T2DM)患者肠道菌群变化的临床意义。方法 选取8例T2DM患者与10例体检健康者,提取其粪便基因组DNA,应用菌属16S rRNA V4序列特异性引物,采用高通量测序技术分析肠道菌群多样性(以香农多样性指数表示)及种类(以丰度表示)。观察不同性别T2DM患者的肠道菌群种类差异,分析T2DM患者肠道优势菌属与空腹血糖(FPG)、餐后1 h血糖(1 h PG)和餐后2 h血糖(2 h PG)的关系。结果 T2DM患者与体检健康者香农多样性指数分别为3.70±0.29、3.30±0.20,二者比较P>0.05。门水平下T2DM患者和体检健康者的肠道共同优势菌门为厚壁菌门、拟杆菌门、变形菌门和放线菌门,二者上述4种优势菌门丰度比较差异均无统计学意义(P均>0.05)。属水平下T2DM患者和体检健康者的肠道共同优势菌属为拟杆菌属和毛螺杆菌科,体检健康者罗斯氏菌属丰度高于T2DM患者(P<0.05)。女性T2DM患者拟杆菌属科丰度高于男性患者,假单胞菌属丰度低于男性患者(P均<0.05)。T2DM患者柔嫩梭菌属丰度与2 h PG呈正相关(r=0.75,P<0.05),其他肠道优势菌属与FPG、1 h PG、2 h PG均无关(P均>0.05)。结论 T2DM患者肠道内存在菌群失调,并具有性别差异;柔嫩梭菌属与血糖水平有关,可为T2DM的治疗提供一种新的思路。
2型糖尿病;肠道菌群;肠道优势菌属;丰度;血糖;高通量测序
研究发现,2型糖尿病(T2DM)的发生不仅与人类基因组差异、饮食结构改变及运动量减少有关,亦与肠道菌群改变有关[1]。T2DM患者多伴有糖脂代谢紊乱,而肠道菌群与宿主之间在调节能量平衡方面联系密切。高通量测序技术多用于检测肠道菌群的结构及多样性,具有快速、准确等特点[2]。本研究采用高通量测序技术观察T2DM患者的肠道菌群结构及多样性变化,现分析结果并探讨其意义。
1 资料与方法
1.1 临床资料 选择2015年9月江苏大学附属医院收治的T2DM患者8例,男4例、女4例,年龄40~70岁。患者均符合WHO关于T2DM的诊断标准[2],排除妊娠糖尿病、特殊类型糖尿病以及合并其他系统性疾病者。选择同期体检健康者10例,男5例、女5例,年龄40~70岁。研究对象采样前3个月内未服用抗生素,近2周内未服用益生元、益生菌等制品,近3个月内无腹泻、便秘、痢疾等胃肠道疾病。
1.2 肠道菌群检测 采用高通量测序技术。受试者于环境温度不高于10 ℃的条件下,采集洁净粪便于储存盒内,并进行密封。于2级生物安全柜内称取0.25 g粪便样本,2 h内送到实验室。采用PowerSoil®-htp 96 Well Soil DNA Isolation Kit提取试剂盒(美国MoBio公司),根据试剂盒说明书提取粪便基因组DNA,检测其质量合格后于-20 ℃保存。利用带有barcode的特异性引物扩增16S rDNA的V4区域,并进行精确定量和纯化回收。构建好的文库经过精确定量和Agilent 2200质检合格后,采用Illumina v3 kit(2×300)于MiSeq(美国Illumina公司)上机检测肠道菌群。方法如下:采用微生物16S rRNA分析管道(QIIME)将优质序列聚类成操作分类单元(OTUs),并对所有样品的优质序列按0.97的相似度进行OTUs聚类,选取每一类中最长的序列作为其代表序列。采用RDP-classifier对OTUs代表序列进行注释,得到每个OTUs的分类学信息。统计各个样品中的OTUs分类数量,应用Mothur软件(version 1.36.0) 计算针对单个样品物种多样性分析的香农多样性指数。香农多样性指数越大,说明样品中的物种越丰富[3]。于门水平及属水平下对OTUs数据进行汇总,并计算门水平和属水平下的菌群丰度(即该菌群在总体菌群中的比例,以百分率表示)。对不同性别T2DM患者的OTUs进行分类汇总,计算肠道菌群丰度。记录T2DM患者的肠道优势菌属,分析其丰度与患者空腹血糖(FPG)、餐后1 h血糖(1 h PG)和餐后2 h血糖(2 h PG)的关系。
1.3 统计学方法 采用SPSS21.0统计软件。计量资料以四分位数法表示,结果比较采用Mann-WhitneyU检验;相关性分析采用Spearman相关分析法。P<0.05为差异有统计学意义。
2 结果
2.1 OTUs分类数量及多样性 T2DM患者与体检健康者OTUs分类数量分别为447.50±36.78、386.60±15.26,香农多样性指数分别为3.70±0.29、3.30±0.20,二者比较P均>0.05。
2.2 门水平下的肠道菌群结构 门水平下T2DM患者和体检健康者的肠道共同优势菌门为厚壁菌门、拟杆菌门、变形菌门和放线菌门,二者上述4种优势菌门丰度比较差异均无统计学意义(P均>0.05)。见表1。
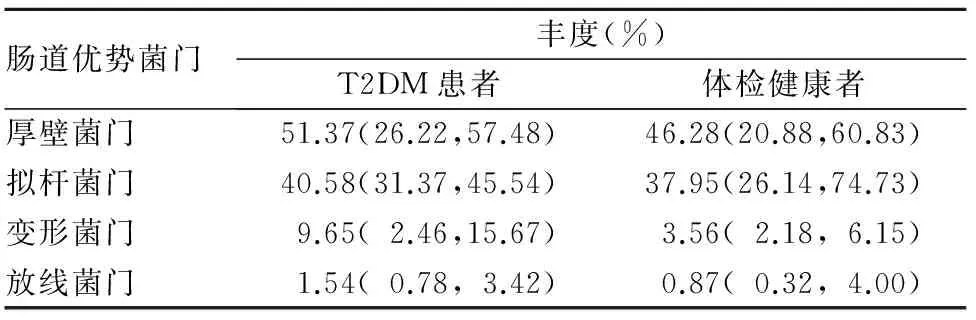
表1 T2DM患者和体检健康者门水平下的肠道优势菌门丰度[M(P25,P75)]
2.3 属水平下的肠道菌群结构 属水平下T2DM患者的肠道优势菌属为拟杆菌属、毛螺杆菌科、瘤胃球菌科和埃希氏菌属等,体检健康者的肠道优势菌属为普雷沃氏菌属、拟杆菌属、毛螺杆菌科和Blautia等。拟杆菌属和毛螺杆菌科是T2DM患者和体检健康者的肠道共同优势菌属,体检健康者罗斯氏菌属丰度高于T2DM患者(P<0.05)。见表2。
2.4 不同性别T2DM患者的肠道菌群结构 男性T2DM患者的肠道优势菌群为拟杆菌属、瘤胃球菌科、毛螺杆菌科、埃希氏菌属、梭菌目、普雷沃氏菌属等,女性患者的肠道优势菌群为拟杆菌属、毛螺杆菌科、埃希氏菌属等。拟杆菌属、毛螺杆菌科和埃希氏菌属为男女性T2DM患者的肠道共同优势菌群。女性T2DM患者拟杆菌属丰度高于男性患者,假单胞菌属丰度低于男性患者(P<0.05)。见表3。
2.5 T2DM患者血糖与肠道优势菌属丰度的关系 T2DM患者柔嫩梭菌属丰度与2 h PG呈正相关(r=0.75,P<0.05),与FPG、1 h PG均无关(P均>0.05)。T2DM患者其他肠道优势菌属丰度与FPG、1 h PG、2 h PG均无关(P均>0.05)。见表4。
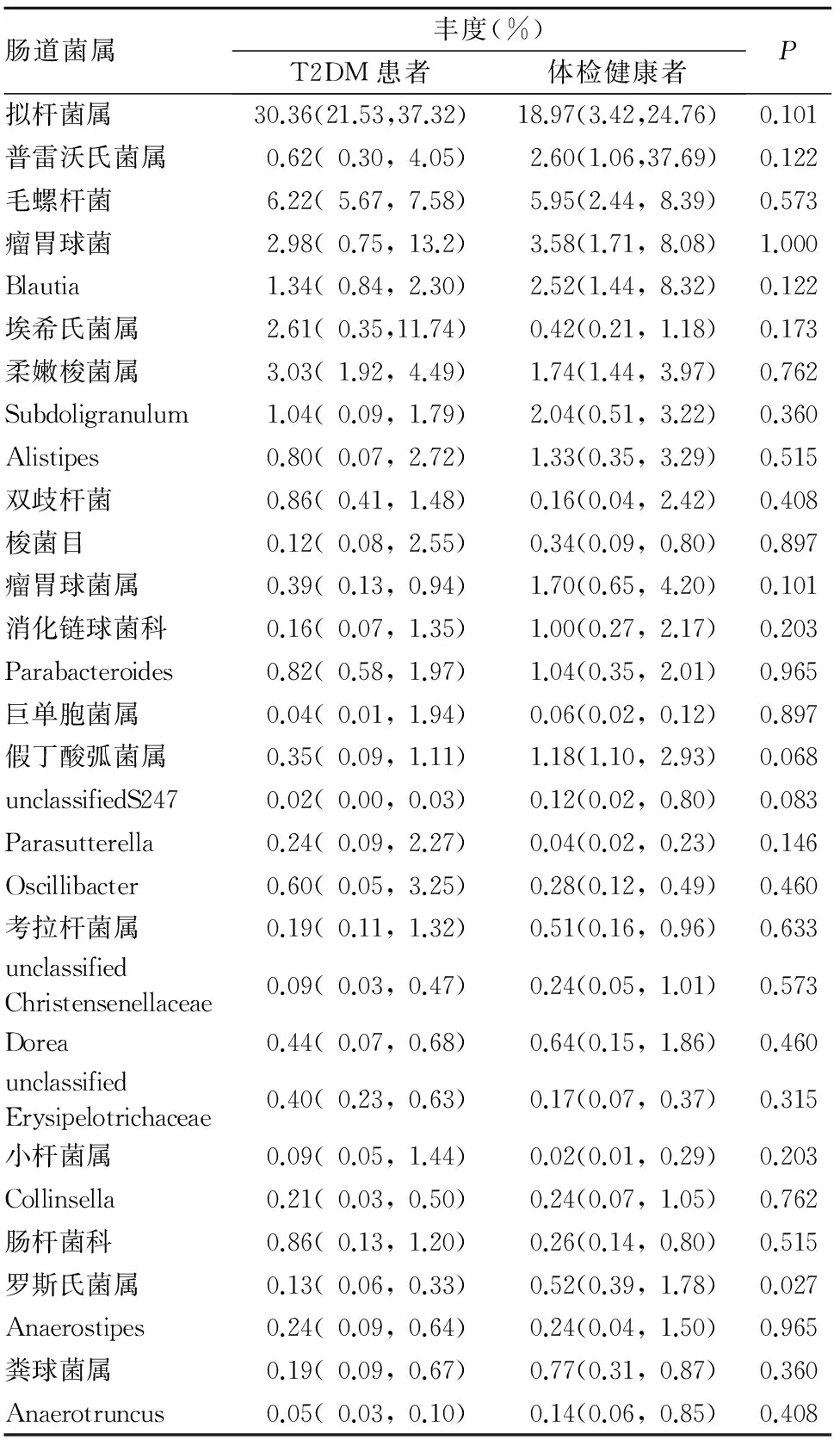
表2 T2DM患者和体检健康者属水平下的肠道优势菌属丰度比较[M(P25,P75)]
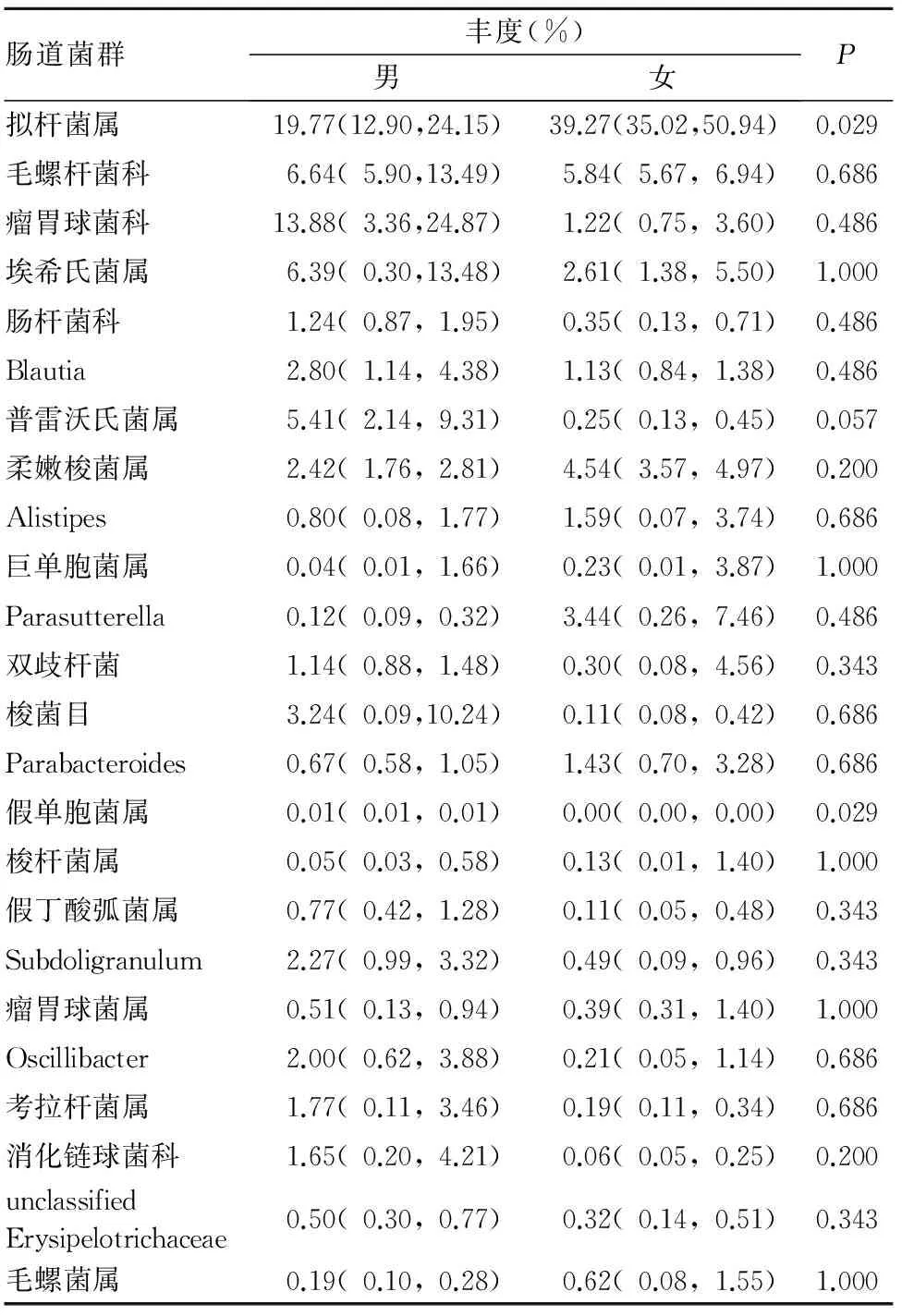
表3 不同性别T2DM患者的肠道菌群丰度比较[M(P25,P75)]
3 讨论
肠道菌群是人体微环境的重要组成部分, 也是最大、最复杂的微生态系统,其多样性可能是宿主和肠道菌群之间强烈选择和协同进化的结果[4]。肠道中的正常菌群能够为宿主提供或合成一些营养要素,如B族维生素、叶酸、生物素等[5]。随着研究的深入,人们逐渐发现,肠道菌群不仅与营养相关,也与人体健康和疾病发生有着极为密切的联系。肠道正常菌群与肠黏膜紧密结合,构成肠道的生物屏障,能够通过营养竞争、占位效应及其代谢产物等抑制条件致病菌的过度生长和外来致病菌的入侵,从而维持肠道菌群平衡[6]。研究表明,肠道菌群与肥胖[7,8]、糖尿病[9]、心脑血管疾病[10,11]和肿瘤[12]等疾病的发生密切相关。
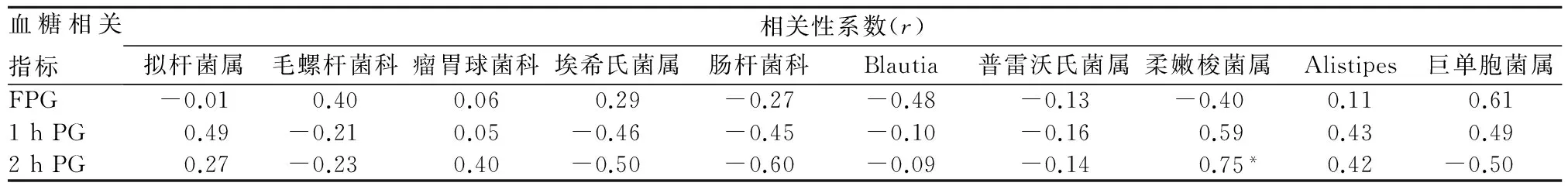
表4 T2DM患者血糖相关指标与肠道优势菌属丰度的关系
注:*P<0.05。
目前临床采用传统培养法培养的细菌仅占肠道菌群的1%~10%,并不能反映菌群的多样性[13];变性梯度凝胶电泳、荧光定量PCR技术也只能检测出一定丰度以上的细菌或特定微生物[14,15]。高通量测序技术又称下一代测序技术,是在第一代Sanger测序基础上发展而来的新兴测序技术,能够一次性对高达数百万条的DNA序列进行测定,使得对菌群进行全面细致的分析成为可能。以Illumina为测序平台的测序技术在研究人类肠道宏基因组方面发挥重要作用[5,16]。与传统培养方法和变性梯度凝胶电泳等分子生物学方法不同,高通量测序技术能更全面地反映微生物群体的物种组成、分布及丰度信息。
Karlsson等[17]研究发现,糖尿病患者肠道菌群的结构、功能与血糖、糖耐量异常均有关。 Larsen等[18]研究发现,与正常人相比,T2DM患者粪便中β-变形菌丰度明显升高,拟杆菌门与硬壁菌门的比值以及普氏拟杆菌与肠球菌的比值与血糖水平呈正相关。本研究发现,T2DM患者与健康人群门水平的4种优势菌门丰度比较差异均无统计学意义;但T2DM患者属水平肠道菌群的丰度与健康人群存在一定的差异,尤其是可以产生丁酸的罗斯氏菌属丰度显著低于健康人群。丁酸是肠道上皮细胞重要的能量来源,菌罗斯氏菌、Blautia属、假丁酸弧菌属和柔嫩梭菌都是人肠道内重要的丁酸产生菌。Qin等[19]研究发现,丁酸产生菌是区别健康人群与T2DM患者的重要菌群。本研究还发现,T2DM患者柔嫩梭菌属丰度与2 h PG呈正相关,进一步验证了上述结论。本研究女性T2DM患者肠道中的优势菌属明显少于男性患者,拟杆菌属丰度高于男性患者,假单胞菌属丰度低于男性患者,说明男女性T2DM患者的肠道菌群存在差异。以上结果均提示,肠道菌群在T2DM的发生、发展中具有一定作用,但是具体机制尚不清楚;虽然男女性T2DM患者血糖均升高,但其升高的机制可能存在差异。本研究T2DM患者门水平下变形菌门丰度有高于健康人群的趋势,属水平下Blautia和假丁酸弧菌属丰度有低于健康人群的趋势,拟杆菌属和埃希氏菌属丰度有高于健康人群的趋势,但均无统计学差异,可能与本研究纳入例数较少有关,仍需扩大样本进一步验证。
综上所述,T2DM患者肠道内存在菌群失调,并具有性别差异;柔嫩梭菌属与血糖水平有关,可为T2DM的治疗提供一种新的思路。
[1] Cani PD, Geurts L, Matamoros S, et al. Glucose metabolism: focus on gut microbiota, the endocannabinoid system and beyond [J]. Diabetes Metab, 2014,40(4):246-257.
[2] Haraszthy VI, Zambon JJ, Sreenivasan PK, et al. Identification of oral bacterial species associated with halitosis [J]. J Am Dent Assoc, 2007,138(8):1113-1120.
[3] Kemp PF, Aller JY. Bacterial diversity in aquatic and other environments: what 16s rdna libraries can tell us[J] . FEMS Microbiol Ecol, 2004,47(2):161-177.
[4] Isberg RR, Barnes P. Dancing with the host; flow-dependent bacterial adhesion[J]. Cell, 2002,110(1):1-4.
[5] Hooper LV, Midtvedt T, Gordon JI. How host-microbial interactions shape the nutrient environment of the mammalian intestine[J]. Annu Rev Nutr, 2002(22):283-307.
[6] 王友湘,陈庆森.益生菌和肠道黏膜免疫关系的研究进展[J].食品科学,2007,28(8):537-542.
[7] Turnbaugh PJ, Hamady M, Yatsunenko T, et al. A core gut microbiome in obese and lean twins[J]. Nature, 2009,457(7228):480-484.
[8] Ley RE. Obesity and the human microbiome[J]. Curr Opin Gastroenterol, 2010,26(1):5-11.
[9] Markle JG, Frank DN, Mortin-Toth S, et al. Sex differences in the gut microbiome drive hormone-dependent regulation of autoimmunity[J]. Science, 2013,339(6123):1084-1088.
[10] Wang Z, Klipfell E, Bennett BJ, et al. Gut flora metabolism of phosphatidylcholine promotes cardiovascular disease[J]. Nature, 2011,472(7341):57-63.
[11] Collins SM, Surette M, Bercik P. The interplay between the intestinal microbiota and the brain[J]. Nat Rev Microbiol, 2012,10(11):735-742.
[12] Fox JG, Feng Y, Theve EJ, et al. Gut microbes define liver cancer risk in mice exposed to chemical and viral transgenic hepatocarcinogens[J]. Gut, 2010,59(1):88-97.
[13] Stewart CJ, Marrs EC, Magorrian S, et al. The preterm gut microbiota: changes associated with necrotizing enterocolitis and infection[J]. Acta Paediatr, 2012,101(11):1121-1127.
[14] Smith B, Bode S, Skov TH, et al. Investigation of the early intestinal microflora in premature infants with/without necrotizing enterocolitis using two different methods[J]. Pediatr Res, 2012,71(1):115-120.
[15] Madan JC, Salari RC, Saxena D, et al. Gut microbial colonisation in premature neonates predicts neonatal sepsis[J] . Arch Dis Child Fetal Neonatal Ed, 2012,97(6):456-462.
[16] Qin J, Li R, Raes J, et al. A human gut microbial gene catalogue established by metagenomic sequencing[J]. Nature, 2010,464(7285):59-65.
[17] Karlsson FH, Tremaroli V, Nookaew I, et al. Gut metagenome in european women with normal, impaired and diabetic glucose control[J]. Nature, 2013,498(7452):99-103.
[18] Larsen N, Vogensen FK, van den Berg FW, et al. Gut microbiota in human adults with type 2 diabetes differs from non-diabetic adults[J]. PLoS One, 2010,5(2):e9085.
[19] Qin J, Li Y, Cai Z, et al. A metagenome-wide association study of gut microbiota in type 2 diabetes[J]. Nature, 2012,490 (7418):55-60.
Variation of intestinal flora in patients with type 2 diabetes mellitus and the significance
MASuxian1,ZHANGRuirui,WANGSu,DENGJialiang,YANGLu,YUANGuoyue
(1AffiliatedHospitalofJiangsuUniversity,Zhenjiang212001,China)
Objective To detect the variation of intestinal flora in patients with type 2 diabetes mellitus (T2DM) and the clinical significance. Methods The fecal samples were collected from 8 T2DM patients and 10 healthy participants. Total DNA was extracted from stool samples and submitted to high-throughput sequencing with primers targeting V4 region of the 16s rRNA gene. High-throughput sequencing was used to analyze the abundance and diversity of fecal microbiota. The difference of intestinal flora in patients with different gender, and the relationship between the dominant bacteria in the T2DM patients and fasting plasma glucose (FPG), 1 hour postprandial blood glucose (1h PG) and 2 hour postprandial blood glucose (2h PG) were investigated. Results The Shannon diversity index of T2DM patients and healthy subjects was 3.70±0.29 and 3.30±0.20, respectively, and there was no significant difference between the two groups (P>0.05). The dominant bacteria in the two groups were Firmicutes, Bacteroides, Proteobacteria, Actinobacteria at phylum level, and there was no significant difference in the abundance between the two groups (allP>0.05). In addition, the dominant bacteria were Bacteroides and Lachnospiraceae in the two groups at genus level. Compared with healthy subjects, the abundance of Roseburia was significantly reduced in the T2DM patients (P<0.05). The abundance of the Bacteroides in the female T2DM patients was much higher than that of the male patients, while the abundance of Peptostreptococcaceae was much lower than that of the male patients (P<0.05). The abundance of Faecalibacterium was positively correlated with 2 h PG (r=0.75,P<0.05), and the other dominant bacteria were not correlated with FPG, 1 h PG, 2 h PG (allP>0.05). Conclusion T2DM patients show intestinal flora imbalance with gender differences, and the abundance of Faecalibacterium in T2DM patients is related to blood glucose, which may provide a new therapeutic approach for T2DM.
type 2 diabetes mellitus; intestinal flora; intestinal dominant fungi; abundance; blood glucose; high-throughput sequencing
国家自然科学基金面上项目(81570721、81370965);江苏省自然科学基金资助项目(BK20151331);“六大人才高峰”第十二批高层次人才项目(2015-WSN-006);镇江市科技支撑-社会发展重点项目(SH2015028)。
马苏娴(1991-),女,硕士研究生在读,研究方向为内分泌代谢性疾病的诊断与治疗。E-mail: 839282375@qq.com
袁国跃(1971-),男,主任医师,研究方向为内分泌代谢性疾病的诊断与治疗。E-mail: yuanguoyue@hotmail.com
10.3969/j.issn.1002-266X.2017.16.006
R587.1
A
1002-266X(2017)16-0020-04
2016-07-15)
