超高效液相色谱-三重四极杆质谱法测定食品中34种非法添加减肥类化合物
2017-06-15张静娴毛秀红
胡 青, 孙 健, 冯 睿, 张 甦, 于 泓, 张静娴, 毛秀红, 季 申
(上海市食品药品检验所, 上海 201203)
研究论文
超高效液相色谱-三重四极杆质谱法测定食品中34种非法添加减肥类化合物
胡 青, 孙 健, 冯 睿, 张 甦, 于 泓, 张静娴, 毛秀红, 季 申*
(上海市食品药品检验所, 上海 201203)
建立了超高效液相色谱-三重四极杆质谱测定食品(含保健食品)中34种减肥类非法添加化合物的方法。采用Waters CORTECS T3色谱柱(100 mm×2.1 mm, 2.7 μm),以0.1%(v/v)甲酸水溶液-含0.1%(v/v)甲酸的乙腈溶液为流动相,梯度洗脱,在电喷雾离子源、正离子或负离子模式下以多反应监测(MRM)方式检测。西布曲明等29种化合物在0.5~10 μg/L范围内、氯噻嗪等5种化合物在2.5~50 μg/L范围内均呈良好的线性关系,相关系数(r)均大于0.99;西布曲明等29种化合物在5、10和20 μg/kg添加水平下的平均加标回收率为49.2%~136.2%,相对标准偏差(RSD)为0.7%~15.0%(n=6),氯噻嗪等5种化合物在25、50和100 μg/kg添加水平下的平均加标回收率为51.5%~130.9%, RSD为0.8%~14.0%(n=6);西布曲明等29种化合物的检出限为5 μg/kg,定量限为10 μg/kg,氯噻嗪等5种化合物的检出限为25 μg/kg,定量限为50 μg/kg。本方法已应用于实际样品的测定,共检出了12种化合物,有效打击了非法添加行为。
超高效液相色谱-三重四极杆质谱;非法添加;减肥类化合物;食品
近年来,具有减肥降脂功效的食品和保健食品层出不穷,为了达到快速起效的目的,市场上存在部分食品和保健食品中非法添加具有相关功效化学药品的情况[1,2]。过多地服用化学药品易对人们的健康和生命造成威胁,因此减肥食品中化学药品的非法添加问题已受到人们的广泛关注。
与减肥功效相关的化合物主要包括抑制食欲类(如西布曲明及其衍生物等)、中枢兴奋类(安非他明和麻黄碱等)、泻下类和降脂类等。目前,国内外关于非法添加化合物的检测方法主要有显色法[3]、薄层色谱法[4]、高效液相色谱法[5]、高效液相色谱-离子阱质谱法[6,7]、超高效液相色谱-串联质谱法[8-11]、超高效液相色谱-高分辨质谱法[12]、液相色谱-核磁共振法[13]、酶联免疫吸附法[14]等。显色法和薄层色谱法一般作为快速检测的方法;高效液相色谱法在定性方面有所欠缺;液相色谱-核磁共振法操作难度较大,仪器普及率低;酶联免疫吸附法无法对不同类型的化合物同时检测。超高效液相色谱-串联质谱法可实现多种化合物同时定性、定量分析,检测灵敏度高,方法适用性强,易于推广。
我国与减肥类保健食品非法添加相关的检测标准有3个:国家药品检验补充检验方法检验项目批准件2006004、2012005和食药监办许[2010]114号文附件2。检测标准中涉及的化合物只有8个:西布曲明、N-双去甲基西布曲明、N-单去甲基西布曲明、麻黄碱、芬氟拉明、呋塞米、酚酞和咖啡因。因此建立简便易行,灵敏度高,专属性强,涵盖范围广的检测方法以支撑目前市场监管需求已迫在眉睫。
本研究基于国内外文献[15-17]和十多年市场监管检测出的阳性化合物,确定了34种非法添加的减肥类相关的化合物。本研究综合考虑提取效率和成本等因素,采用甲醇超声提取,建立了超高效液相色谱-三重四极杆质谱同时检测食品中34种非法添加减肥类化合物的方法,实现了复杂基质中多组分的同时定性、定量分析,满足复杂基质的高灵敏度检测要求。
1 实验部分
1.1 仪器与试剂
Agilent 1290超高效液相色谱仪;Agilent 6495三重四极杆质谱;B3500S-MT超声波发生器(上海必能信超声有限公司); Waters CORTECS T3色谱柱(100 mm×2.1 mm, 2.7 μm)。
34种对照品的来源及相关信息见表1;乙腈、甲酸(色谱纯,德国Merck公司);甲醇(分析纯,上海凌峰化学试剂有限公司)。

表 1 34种化合物对照品的信息
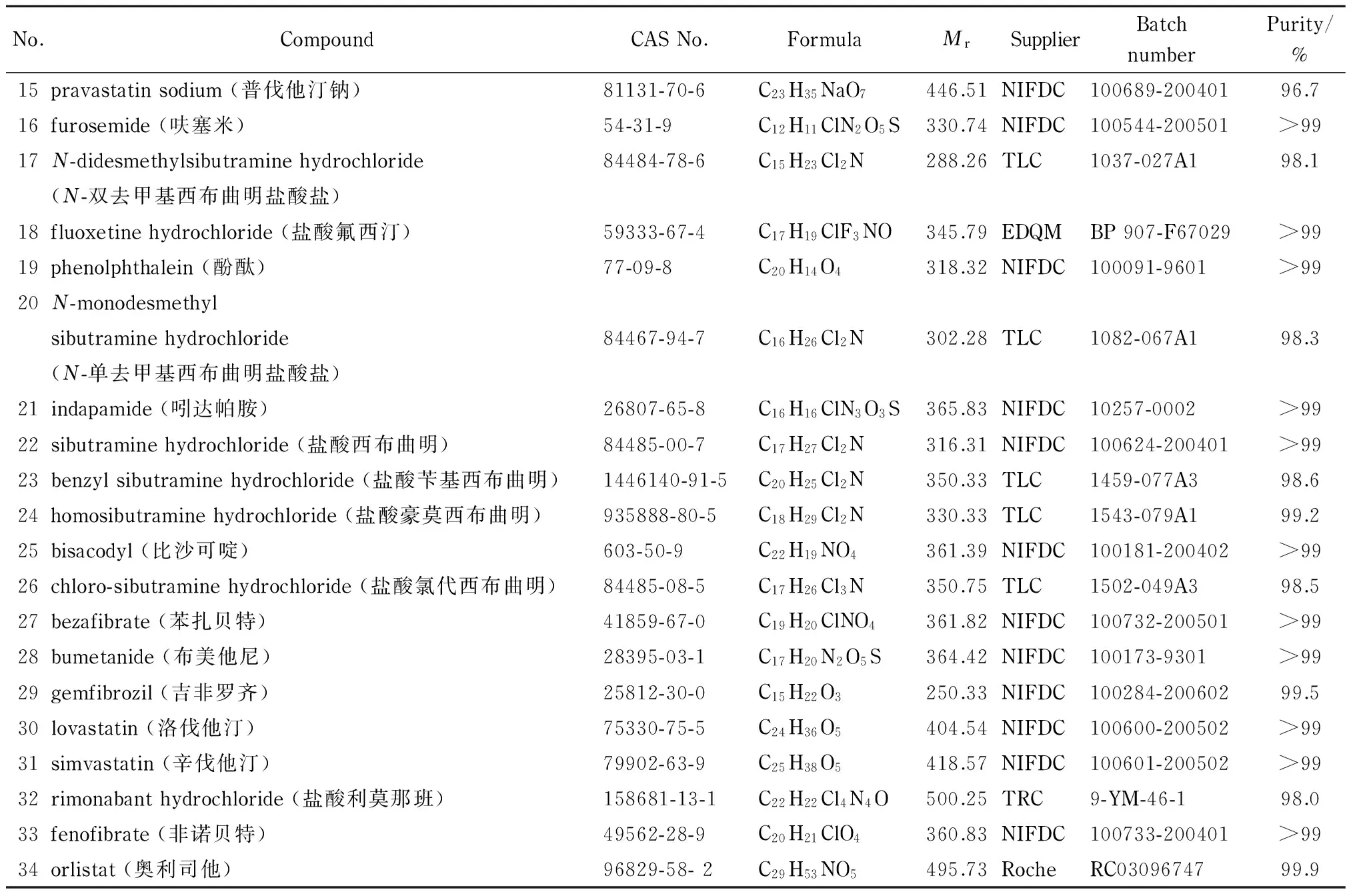
表 1 (续)
NIFDC: National Institutes for Food and Drug Control; TRC: Toronto Research Chemicals Inc.; USP: the United States Pharmacopeial Convention; EDQM: European Directorate for the Quality of Medicines & Healthcare; TLC: TLC Pharmaceutical Standards Ltd.
1.2 分析条件
色谱柱:Waters CORTECS T3 (100 mm×2.1 mm, 2.7 μm);柱温:30 ℃;流动相:A为0.1%(v/v)甲酸水溶液,B为含0.1%(v/v)甲酸的乙腈溶液;梯度洗脱:0~5.0 min,95%A;5.0~22.0 min,95%A~2%A;22.0~27.0 min,2%A;27.0~27.5 min,2%A~95%A;27.5~32.0 min,95%A;流速:300 μL/min;进样量:1 μL。
离子源:电喷雾离子(ESI)源,正离子或负离子模式;离子源温度:200 ℃;毛细管电压:正离子模式4 000 V,负离子模式3 500 V;干燥气流量:12 L/min;雾化气压力:172 kPa;鞘气温度:250 ℃;鞘气(N2)流量:10 L/min;喷嘴电压:正离子模式500 V,负离子模式2 000 V;采用多反应监测(MRM)方式检测。34种化合物的母离子、子离子和碰撞能量(CE)见表2。

表 2 34种化合物的母离子、子离子和碰撞能量
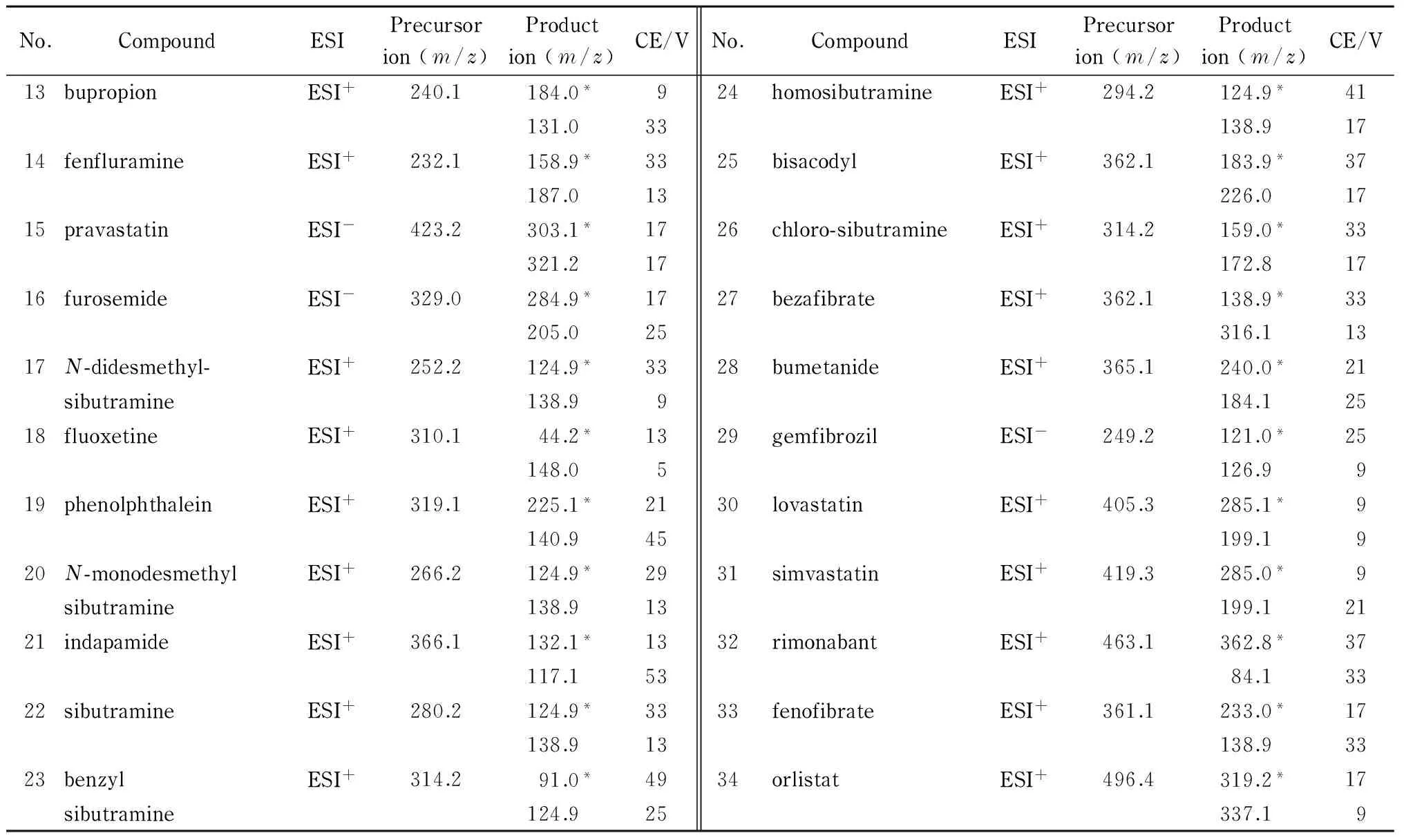
表 2 (续)
* Quantitative ion.
1.3 标准溶液的配制
分别取氯噻嗪、氢氯噻嗪、普伐他汀钠、呋塞米、吉非罗齐对照品(A组)适量,精密称定,用甲醇溶解并配制质量浓度为2.5、5、10、25、50 μg/L的系列混合对照品溶液A,在负离子模式下检测;分别取其他29种对照品(B组)适量,精密称定,用甲醇溶解并配制质量浓度为0.5、1、2、5、10 μg/L的系列混合对照品溶液B,在正离子模式下检测。
1.4 样品前处理
取适量固体样品混匀,研细,准确称取1.000 g粉末,置于具塞试管中,加入10 mL甲醇,密塞,称重,超声提取10 min,放冷至室温后,再次称重,用甲醇补足减失的重量,摇匀,用0.22 μm微孔滤膜(有机相型)过滤,收集续滤液,作为供试品溶液备用。
取适量液体样品摇匀,吸取1.0 mL,置于具塞试管中,加入9 mL甲醇,密塞,称重,超声提取10 min,放冷至室温后,再次称重,用甲醇补足减失的重量,摇匀,用0.22 μm微孔滤膜(有机相型)过滤,收集续滤液作为供试品溶液,备用。
2 结果与讨论
2.1 前处理条件的优化
由于甲醇对待测化合物的溶解性均较好,故以甲醇为提取溶剂进行超声提取。本实验比较了不同超声时间(5、10、15 min)对待测化合物提取效率的影响,结果表明,超声10 min时,待测化合物的提取效率较高,且可以避免基质中干扰成分的共流出。在实际样品测定中,宜根据样品添加量的实际浓度,进行适当的稀释,避免进样浓度过高,对仪器造成污染。2.2 色谱条件的优化
本研究曾比较了分析时间为25、28和32 min时目标化合物的分离效果。结果表明,随着分析时间的延长,目标化合物与干扰基质共流出的情况有所减少,综合考虑检测效率、分离效果等情况,采用梯度洗脱,将分析时间设定为32 min。
本研究比较了0.1%(v/v)甲酸水溶液-含0.1%(v/v)甲酸的乙腈溶液、10 mmol/L甲酸铵溶液-乙腈和0.1% (v/v)甲酸水溶液(含10 mmol/L乙酸铵)-乙腈作为流动相时目标化合物的分离效果。结果表明,以0.1%(v/v)甲酸水溶液-含0.1%(v/v)甲酸的乙腈溶液为流动相时,各待测化合物响应好,灵敏度高,离子化效率最佳。 目标化合物中苯丙醇胺等化合物的极性较大,保留时间较短,因此,0~5.0 min时,流动相采用高比例的水相,以延缓其出峰时间,并且能将两组出峰较快的同分异构体(苯丙醇胺和去甲伪麻黄碱、麻黄碱和伪麻黄碱)较好的分离;非诺贝特、奥利司他等化合物的非极性较强,15 min后采用较高比例的有机相洗脱,为尽可能减少在较高有机相比例洗脱时待测化合物与复杂基质中干扰化合物共流出的情况,将有机相的比例设定为逐渐升高;最终以98%(v/v)的有机相冲洗色谱柱,再恢复到初始流动相比例。
2.3 质谱条件的优化
分别用甲醇溶解34种化合物,并配制质量浓度为0.5 mg/L的标准溶液,使用单针自动进样的方式分析目标化合物。分别在正、负离子模式下进行全扫描,采用Agilent MassHunter Optimizer软件自动筛选定量及定性离子对,并对碰撞能量进行优化。将分子离子作为母离子,并给予一定的碰撞能量,对二级离子进行全扫描,选取相对丰度较高的2个子离子分别作为定量和定性离子,并在MRM模式下进一步优化碰撞能量、碎裂电压等质谱参数。氯噻嗪、氢氯噻嗪、普伐他汀、呋塞米、吉非罗齐5种化合物在负离子模式下的响应优于正离子模式下的响应,因此上述5种化合物在ESI-模式下检测;其余29种化合物在ESI+模式下检测。34种化合物的总离子流色谱图分别见图1a和图1b。
研究中发现,氟西汀的碎片离子中m/z大于50的只有m/z为148.0的碎片离子,通过扩大相对分子质量的扫描范围和参考相关文献[16],发现了另一个响应较好而且稳定的碎片离子(m/z44.2),并且比m/z为148.0的碎片离子响应强度更强。综合考虑,采用m/z为44.2的碎片离子作为氟西汀的定量离子。
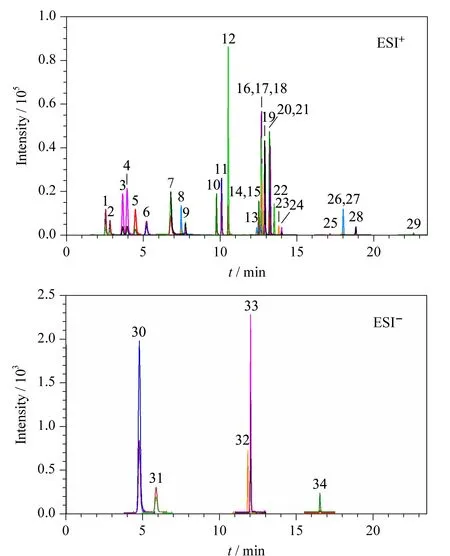
图 1 不同模式下34种化合物的总离子流色谱图Fig. 1 Total ion chromatograms of the 34 compounds 1. phenylpropanolamine; 2. norpseudoephedrine; 3. ephedrine; 4. pseudoephedrine; 5. methylephedrine; 6. amphetamine; 7. methamphetamine; 8. caffeine; 9. phentermine; 10. lorcaserin; 11. bupropion; 12. fenfluramine; 13. indapamide; 14. phenolphthalein; 15. N-didesmethylsibutramine; 16. fluoxetine; 17. N-monodesmethyl sibutramine; 18. bisacodyl; 19. sibutramine; 20. benzyl sibutramine; 21. homosibutramine; 22. chlorosibutramine; 23. bezafibrate; 24. bumetanide; 25. lovastatin; 26. simvastatin; 27. rimonabant; 28. fenofibrate; 29. orlistat; 30. chlorothiazide; 31. hydrochlorothiazide; 32. pravastatin; 33. furosemide; 34. gemfibrozil.
2.4 基质效应的考察
根据市场上常见的减肥产品,选择了酵素梅、左旋肉碱泡腾片、减肥胶囊、酵素饮品、代餐饼干、减肥奶茶、减肥茶、左旋肉碱咖啡粉8种基质作为代表性基质。研究发现,因咖啡粉和减肥茶中存在大量固有成分咖啡因,导致多种化合物的回收率偏差较大。以减肥茶为例,配制基质对照后对化合物进行回收率考察,结果表明,多种化合物的回收率均得到了明显的改善,说明采用空白基质提取液配制系列标准工作溶液可以减小基质效应,从而得到更准确的定量结果。因此在实际检测中遇到如咖啡粉或减肥茶等复杂基质,在可以获得类似空白基质的前提下,使用基质对照,可以更加准确地对目标化合物进行定量。
2.5 方法学验证
2.5.1 仪器精密度
取各对照品溶液(29种化合物为1 μg/L, 5种化合物为5 μg/L),连续进样6次,记录峰面积。结果表明,各化合物峰面积的RSD均低于8%,说明仪器的稳定性良好。
2.5.2 线性关系
对系列混合对照品溶液A和B进行分析,以化合物的峰面积(y)为纵坐标、对应的质量浓度为横坐标(x, μg/L)进行线性回归,结果见表3。结果表明,34种化合物在各自的线性范围内线性关系良好,相关系数(r)均大于0.99。
2.5.3 加标回收率
分别取上述8种基质进行分析,除在酵素饮品、减肥奶茶、减肥茶、左旋肉碱咖啡粉基质中检测到咖啡因外,其余基质中均不含本方法所检测的34种化合物。西布曲明等29种化合物在5、10、20 μg/kg的加标水平下,氯噻嗪等5种化合物在25、50、100 μg/kg的加标水平下进行回收率和精密度试验,每个水平做6个平行样品。西布曲明等29种化合物的平均加标回收率为49.2%~136.2%,相对标准偏差(RSD)为0.7%~15.0%;氯噻嗪等5种化合物的平均加标回收率为51.5%~130.9%, RSD为0.8%~14.0%(见表S1,详见http://www.chrom-China.com/UserFiles/File/1703007-SI.pdf)。

表 3 34种化合物的线性方程和相关系数
y: peak area;x: mass concentration, μg/L.
由于酵素饮品、减肥奶茶、减肥茶、左旋肉碱咖啡粉中均含有咖啡因,故未计算咖啡因的回收率。奥利司他在减肥茶制品中易受到干扰,故针对减肥茶、减肥奶茶、咖啡粉基质仅定性,未计算回收率结果。按照GB/T 27404-2008标准,低、中、高3个水平的加标回收率一般要求为60%~120%,总体来看,回收率结果中有92%的数据位于60%~120%之间,说明所建立的方法定量准确。
2.5.4 稳定性
取供试品溶液,分别于放置0、24、48 h时进样测定,比较各化合物的峰面积。结果表明,34种化合物的RSD为2.1%~15.8%,表明供试品溶液在48 h内基本稳定。
2.5.5 检出限和定量限
向8种基质样品中添加低含量的34种化合物,以本方法可以检出的含量作为检出限,结果表明,正离子模式下,29种化合物的检出限为5 μg/kg(固体)或5 μg/L(液体);负离子模式下,5种化合物的检出限为25 μg/kg(固体)或25 μg/L(液体);以可以准确定量的含量作为定量限,结果表明,正离子模式下,29种化合物的定量限为10 μg/kg(固体)或10 μg/L(液体);在负离子模式下,5种化合物的定量限为50 μg/kg(固体)或50 μg/L(液体)。在减肥奶茶、减肥茶、咖啡粉等基质中,奥利司他受基质干扰较严重,回收率普遍偏低,且RSD值较大,故在固体冲饮品基质中,奥利司他仅作定性检测,检出限为100 μg/kg。
为进一步研究检出限能否达到非法添加含量的测定要求,对34种化合物的临床用量进行调研,其中布美他尼的临床用量最少,为0.5 mg/次。如果暂以1次1 g计,则最低起效的药物含量为0.5 mg/g,即上述化合物至少添加0.5 mg/g才能起效。本方法的检出限远远低于0.5 mg/g,符合实际非法添加含量测定的要求。
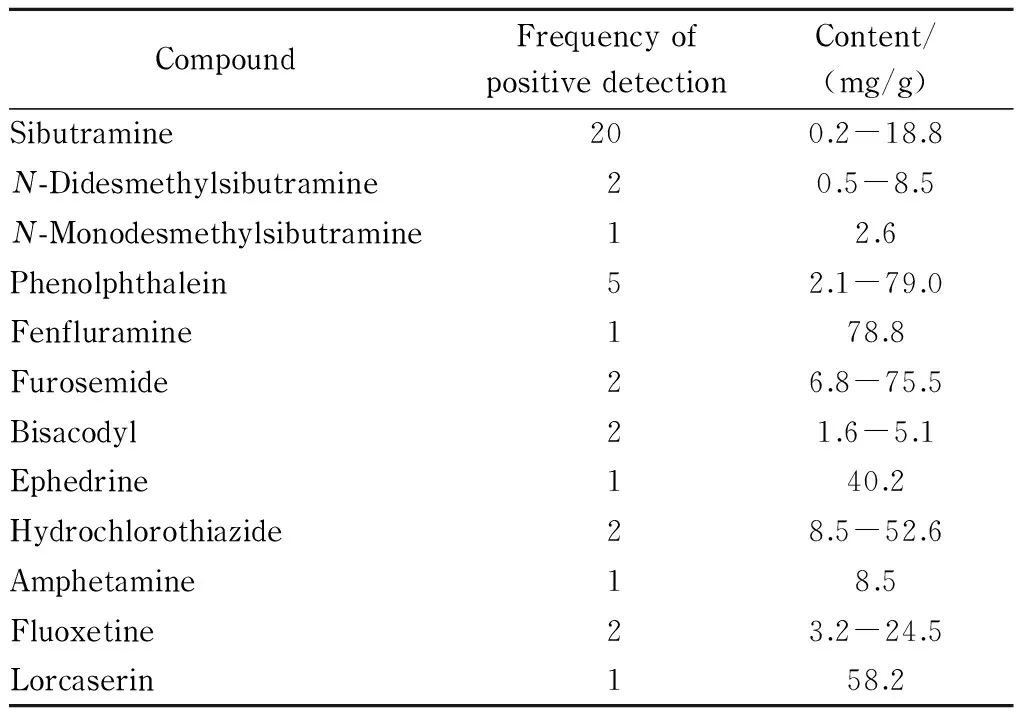
表 4 80批实际样品的测定结果
2.6 实际样品分析
本研究对市售样品及某涉案专项样品(共80批)进行检测,共检出12种化合物,检出频次达40次(见表4)。并有多种化合物同时添加的情况,如同时添加西布曲明与酚酞,且添加含量差异较大,甚至添加含量是临床用量的数倍,表明了非法添加的随意性及不可预料性,对消费者健康具有较大危害。
3 结论
本研究建立了超高效液相色谱-三重四极杆质谱测定食品(含保健食品)中34种减肥类非法添加化合物的分析方法。方法简便、灵敏、准确,现已转化为国家食品补充检验方法,大幅增加了检测非法添加化合物的种类,对打击非法添加保障食药安全提供了有力的技术支持。
[1] Food and Drug Administration. Beware of Products Promising Miracle Weight Loss. (2015-07-25) [2017-04-05]. https: //www.fda.gov/forconsumers/consumerupdates/ucm246742. htm
[2] Wozniak K S, Georgiev M I, Orhan I E. Food Chem Toxicol, 2016, 6: 1
[3] Huang N J, Yang W H, Huang Y B. Food and Drug, 2011, 13(3): 114
黄诺嘉, 杨文红, 黄奕滨. 食品与药品, 2011, 13(3): 114
[4] Wan L C, Wang D, Ding Y P, et al. Chinese Pharmaceutical Affairs, 2013, 27(12): 1285
万林春, 王栋, 丁银平, 等. 中国药事, 2013, 27(12): 1285
[5] Wang J W, Huang X L, Cao J, et al. Chinese Journal of Chromatography, 2014, 32(2): 151
王静文, 黄湘鹭, 曹进, 等. 色谱, 2014, 32(2): 151
[6] Zeng Y, Xu Y M, Kee C L, et al. Drug Test Anal, 2016, 8: 351
[7] Wang K, Hu Q, Cui Y L, et al. Chinese Journal of Pharmaceutical Analysis, 2008, 28(8): 1268
王柯, 胡青, 崔益泠, 等. 药物分析杂志, 2008, 28(8): 1268
[8] Wang J, Chen B, Yao S. Food Addit Contam A, 2008, 25(7): 822
[9] Rebiere H, Guinot P, Civade C, et al. Food Addit Contam A, 2012, 29(2): 161
[10] Kim H J, Lee J H, Park H J, et al. Food Addit Contam A, 2014, 31(5): 777
[11] Liang Q L, Qu J, Luo G A, et al. J Pharm Biomed Anal, 2006, 40: 305
[12] Yu H, Hu Q, Ji S, et al. Journal of Food Safety & Quality, 2016, 7(7): 2704
于泓, 胡青, 季申, 等. 食品安全质量检测学报, 2016, 7(7): 2704
[13] Vaysse J, Balayssac S, Gilard V, et al. Food Addit Contam A, 2010, 27(7): 903
[14] Liu B, Yuan L P, Nie Y H, et al. Modern Food Science and Technology, 2016, 32(8): 290
刘波, 袁利鹏, 聂燕华, 等. 现代食品科技, 2016, 32(8): 290
[15] Lu H, Qian Y F, Zhang B, et al. Science and Technology of Food Industry, 2016, 37(10): 88
鲁辉, 钱叶飞, 张斌, 等. 食品工业科技, 2016, 37(10): 88
[16] Ma W, Peng T, Zhu M D, et al. Chinese Journal of Analytical Chemistry, 2009, 37(11): 1583
马微, 彭涛, 朱明达, 等. 分析化学, 2009, 37(11): 1583
[17] Gong X, Lu L, Feng Y L, et al. Chinese Journal of Pharmaceutical Analysis, 2016, 36(5): 918
宫旭, 芦丽, 冯有龙, 等. 药物分析杂志, 2016, 36(5): 918
Platform Project of Science and Technology Commission of Shanghai Municipality (No. 14DZ2294000); 2016 Food Detection Supplementary Method Development Project of the State Food and Drug Administration.
Determination of 34 illegally adulterated weight loss compounds in foods by ultra high performance liquid chromatography-triple quadrupole mass spectrometry
HU Qing, SUN Jian, FENG Rui, ZHANG Su, YU Hong,ZHANG Jingxian, MAO Xiuhong, JI Shen*
(ShanghaiInstituteforFoodandDrugControl,Shanghai201203,China)
An analytical method was developed for the determination of 34 illegally adulterated weight loss compounds in foods and dietary supplements by ultra high performance liquid chromatography-triple quadrupole mass spectrometry (UHPLC-MS/MS). A Waters CORTECS T3 column (100 mm×2.1 mm, 2.7 μm) was used with 0.1% (v/v) formic acid aqueous solution-acetonitrile containing 0.1% (v/v) formic acid as mobile phases by gradient elution. The compounds were detected by electrospray ion source in positive or negative mode with multiple reaction monitoring (MRM) mode. The calibration curves showed good linearity in the range of 0.5-10 μg/L for 29 compounds such as sibutramine, and in the range of 2.5-50 μg/L for five compounds such as chlorothiazide. The correlation coefficients (r) of the standard calibration curves for the 34 analytes were all greater than 0.99. The recoveries of the 29 compounds at spiked levels of 5, 10 and 20 μg/kg were in the range of 49.2%-136.2%, and the RSDs were 0.7%-15.0% (n=6). The recoveries of the five compounds at spiked levels of 25, 50 and 100 μg/kg were in the range of 51.5%-130.9%, and the RSDs were 0.8%-14.0% (n=6). The limits of detection (LODs) and limits of quantification (LOQs) were 5 μg/kg and 10 μg/kg for the 29 compounds, 25 μg/kg and 50 μg/kg for the five compounds, respectively. The method was successfully applied to the analysis of actual samples, and 12 compounds were checked out, which combated the illegal adulteration behavior effectively.
ultra high performance liquid chromatography-triple quadrupole mass spectrometry (UHPLC-MS/MS); illegal adulteration; weight loss compounds; foods
10.3724/SP.J.1123.2017.03007
2017-03-07
上海市科委技术平台专项(14DZ2294000); 2016年国家食品药品监督管理总局食品补充检验方法项目.
O658
A
1000-8713(2017)06-0594-07
* 通讯联系人.Tel:(021)50798195,E-mail:jishen2013@163.com.
