激光剥蚀电感耦合等离子体质谱法中生物样品的元素分馏效应研究
2017-06-15李青张国霞陈奕睿汪正丁传贤
李青+张国霞+陈奕睿+汪正+丁传贤


摘 要 采用213 nm纳秒激光剥蚀系统对生物基体样品的剥蚀颗粒进行研究,优化了激光剥蚀条件。在剥蚀能量为25%,束斑直径为200 μm,剥蚀速率为20 μm/s,频率为20 Hz,载气为700 mL He + 700 mL Ar时,信號强度及稳定性最佳。以31P为内标元素,最佳剥蚀条件下,考察了56个元素的相对分馏因子。结果表明,生物基体的剥蚀颗粒相较于NIST 610 玻璃标样更大,达到3 μm;生物基体中元素分馏效应相较于玻璃基体小,大多数元素的相对分馏因子达到1.0 ±0.1。探讨了生物基体中元素分馏机理,分析了生物基体相较于玻璃基体剥蚀颗粒大,而相对分馏因子未明显增大的原因。一方面可能是粒径3 μm的颗粒进入电感耦合等离子体后能原子化;另一方面,大的剥蚀颗粒的富集效应相对较小。进一步对分馏效应的影响因素进行研究,发现分馏效应与激光剥蚀能量、激光频率和扫描速率相关,并且与元素的氧化物沸点负相关,与氧化物键能和电离能正相关。
关键词 激光剥蚀电感耦合等离子体质谱; 纳秒激光; 分馏效应; 生物样品
1 引 言
近年来,激光剥蚀电感耦合等离子体质谱法(LAICPMS) 作为一种重要的原位微区分析技术手段,具有空间分辨率高、检出限低等特点,已广泛应用于地球化学、冶金、生物医药等领域[1~4]。尤其是在生物医药领域,研究者采用LAICPMS研究抗癌药物在靶器官中的分布,并将其应用到蛋白组学、金属组学等领域[5,6]。然而LAICPMS仍面临着巨大的挑战——定量校准[4]。为克服这一难题,研究者建立了基体匹配[7]、内标校正[7]、在线溶液校准[8]等方法,但仍难于达到溶液样品进样ICPMS分析定量校准的线性和标准偏差要求,主要原因在于激光物质相互作用的复杂性,例如分馏效应[9]。
分馏效应是指不同元素在剥蚀蒸发及传输过程中行为的差异,测得的样品组分与样品组成有一定差异,即元素的非计量剥蚀。它的基体差异性发生在样品剥蚀、传输、蒸发、原子化和电离过程中。对分馏效应的研究多集中在硅酸盐样品或是金属基质样品[10,11],研究了分馏效应与激光波长、激光脉冲宽度、激光能量、激光束斑及在ICP中的传输和离子化的影响[12,13]。但对于生物样品的分馏效应研究却较少。目前,LAICPMS方法在生物医药领域的广泛应用,使得对生物样品分馏效应的研究显得尤为重要。
本研究以动物组织为研究对象,研究了213 nm纳秒激光剥蚀系统对生物基体样品的剥蚀颗粒,考察了常见元素和微量元素的分馏效应,探讨了元素分馏机理,并对分馏效应的影响因素进行了研究。
2 实验部分
2.1 仪器及参数
采用LSX213型号213 nm激光剥蚀系统(美国Cetac公司)与Thermo X Series Ⅱ电感耦合等离子体质谱仪 (美国 ThermoFisher Scientific公司) 联用的 LAICPMS 系统。ICPMS的主要工作参数选择见表 1。
2.2 试剂
2.3 实验样品
实验所用生物基体样品均为实验室自行配制的标准样品。取猪肾(购于本地超市)切成0.5 cm×0.5 cm薄片,在真空干燥箱中烘干, 待用。配制100和1000 μg/L标准溶液,用移液枪取6.8 μL滴至猪肾切片上,室温下自然风干。对自行配制的标准样品进行线扫描剥蚀,其标准曲线相关系数达到0.99~0.999, 信号RSD<6.5%,认为配制的标准样品具备元素分布均匀性和数据准确性,可用于分馏效应研究。
实验玻璃基体样品为美国标准技术研究院(NIST)合成玻璃标准物质 NIST 610。
2.4 激光剥蚀颗粒的收集及表征
在最佳激光剥蚀条件下剥蚀生物基体样品和玻璃基体样品(NIST610),产生的剥蚀气溶胶通过载气输出,在输出口放置干净硅片并固定,收集30 min剥蚀颗粒后,将硅片密封,待测。利用场发射扫描电子显微镜(JSM6700F,日本电子株式会社)表征激光剥蚀颗粒形貌。
3 结果与讨论
3.1 激光参数优化
为获得最优信号强度及稳定性,以Cr为例,对激光剥蚀能量、束斑直径、扫描速率、频率和载气进行了优化,结果如图1所示。结果表明,当剥蚀能量为25%,束斑直径为200 μm,剥蚀速率为20 μm/s,频率为20 Hz,载气为He + Ar=700 mL+700 mL时,信号强度及RSD最佳。
3.2 分馏效应机理探讨
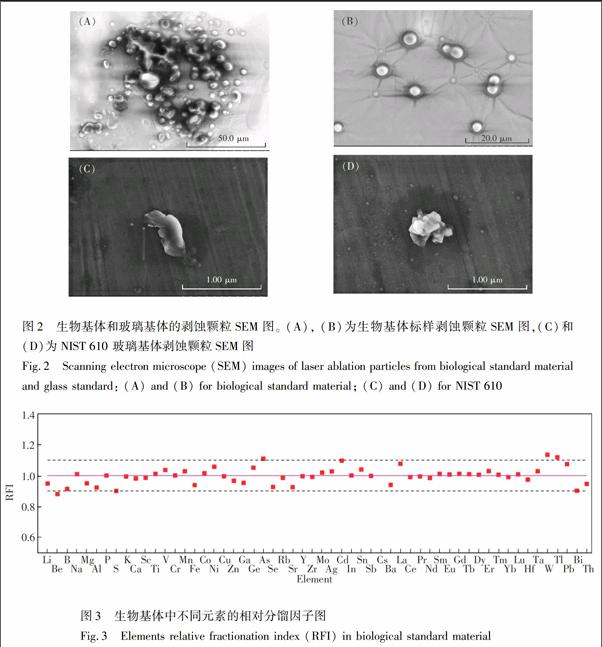
本研究所使用的是纳秒激光剥蚀系统, 相对于飞秒激光剥蚀系统分馏效应更强。这主要是由于纳秒激光的脉冲持续时间较长,通过雪崩电离获得激光能量的电子在脉冲结束前会将能量传递给晶格,因此在一定聚焦体积内,通过热聚焦使样品局部被加热,导致化学键断裂和部分熔融。样品内产生的热梯度导致从聚焦区至周围的热传递扩散,使得样品的剥蚀和熔融区远大于初始聚焦区。正是这种热效应导致样品产生大小不一的颗粒。Kuhn等[15]对玻璃标样的分馏效应进行研究,收集了不同粒径范围的剥蚀颗粒,测定了其中的成分,发现剥蚀颗粒大小对分馏效应有直接影响,粒径小于125 nm和340 nm的剥蚀颗粒中Cu, Zn, Ag, Tl, Pb 和 Bi等易挥发元素相较于Ca出现明显富集;但其中不完全蒸发、原子化、离子化的大颗粒是分馏效应的主要原因[15]。由于激光剥蚀条件和基体会产生不同的剥蚀颗粒,造成不同的元素分馏效应,因此我们对生物基体样品和玻璃基体标样的剥蚀颗粒进行表征,比较了两种不同基体间的分馏效应。 图2为生物基体和玻璃基体标样的剥蚀颗粒SEM表征图。从图2可见,生物基体的剥蚀颗粒约为3 μm,且多为较规则的球形;玻璃基体的剥蚀颗粒小于0.5 μm,其形状多不规则;生物基体的剥蚀颗粒较玻璃标样的大,这可能是由于基质不同导致;生物样品剥蚀的激光能量阈值较低[16],在相同剥蚀条件下,生物样品所受到的激光能量密度超出其阈值,导致能量快速转移进入材料,在其融化的样品表面产生微米级颗粒,同时较大的颗粒也会发生团聚[9,17]。
选取生物基体中31P 元素为内标,在上述优化的最佳剥蚀条件下对大部分ICPMS可测元素相对分馏因子进行考察,结果如图3所示。实验未选用13C为内标元素,是考虑到国内供应的氦气和氩气中13C 的背景通常较高,不宜使用这种方法[18]。结果表明,生物基体中多数元素的分馏因子为1.0 ±0.1。与玻璃基体相比,生物基体中元素分馏因子较小,这可能是由于基体组成元素及元素间的相互作用有关。玻璃基体中,剥蚀产生的颗粒越小,元素分馏效应越大,尤其是易挥发元素(Ag, Cd, In等)受剥蚀颗粒粒径影响更为明显[19]。虽然生物基体剥蚀颗粒的粒径大于玻璃基体,但研究表明粒径小于5 μm的颗粒进入ICP后能够原子化[20];另一方面,根据剥蚀颗粒的SEM表征结果说明生物基体中大剥蚀颗粒较多,而大颗粒的富集效应相对较小,因此生物基体的相对分馏因子相较于玻璃基体更接近1。
3.3 不同激光参数对分馏效应的影响
本研究所用的激光剥蚀能量均为25%,通过在线扫描的模式下,改变束斑直径分别为50、100和200 μm, 考察激光剥蚀能量密度对生物基体样品中V、Cr、Mn、Fe、Co、Ni、Cu、Ag、Cd、Tl、Pb等元素的分馏效应的影响,结果如图4A所示。在束斑直径为200 μm,频率为20、10和5 Hz时考察频率对分馏效应的影响。如图4B所示,激光频率对分馏效应影响较大。随着频率增大,相同时间内剥蚀下来的样品越多,相对分馏因子更接近1.0;当频率降至10 Hz时,相对分馏因子超出了1.0 ±0.1的范围。图4C为激光剥蚀速率分别30,20,10 μm/s对分馏效应的影响,发现随着剥蚀速率的减小,相对分馏因子更接近1。
即束斑直径为200 μm,频率为20 Hz,剥蚀速率为10 μm/s时,相对分馏因子多在1.0附近。难熔元素如V,Co,Fe等的相对分馏因子与易挥发元素(如Tl、Cd、Zn等)的波动较小。随着剥蚀量的增加,元素在剥蚀气溶胶中的富集效应更小,更能体现标样的均匀性,使得相对分馏因子更接近1.0。
进一步对分馏效应的影响因素进行研究,在上述最佳激光剥蚀条件下,考察了分馏效应与电离能、氧化物沸点和键能的关系。从图5可见,元素相对分馏因子和元素氧化物沸点呈负相关,与电离能和氧化物键能呈正相关[20],表明氧化物沸点低的元素容易原子化,其相对分馏因子增大;电离能和氧化物键能低的元素非金属性越強,相应的氧化物熔点越高,同理,在ICP中原子化能力越差,因此其相对分馏因子越小。
4 结 论
本研究以动物组织为研究对象,研究了213 nm纳秒激光剥蚀系统对生物基体样品和玻璃基体标准样品的剥蚀颗粒。形貌表征结果表明,生物基体的剥蚀颗粒相较于NIST 610 玻璃标样更大,达到3 μm。以31P为内标元素,利用相对分馏因子考察了各元素的分馏效应,发现生物基体中元素分馏效应相较于玻璃基体小,大多数元素的相对分馏因子达到1.0 ± 0.1。探讨了生物基体中元素分馏机理,分析了生物基体相比于玻璃基体剥蚀颗粒大,而相对分馏因子未明显增大的原因。一方面可能是粒径3 μm的颗粒进入ICP后仍能原子化;另一方面,大剥蚀颗粒的富集效应相对较小。进一步对分馏效应的影响因素进行研究发现,分馏效应与剥蚀量有直接关系,并且分馏效应与氧化物沸点呈负相关,而与电离能和氧化物键能呈正相关。由于分馏效应不能完全避免,因此在研究时应根据需求对分馏效应、信号强度及稳定性有所取舍。本研究为生物样品LAICPMS法准确定量分析的可行性提供了重要信息。
References
1 GarbeSchnberg D, Müller S. J. Anal. At. Spectrom., 2014, 29(6): 990-1000
2 WANG Ying, GUO YanLi, YUAN HongLin, WEI YongFeng, YAN HongTao, CHEN HuiHui. Spectroscopy and Spectral Analysis, 2012, 32(1): 223-228
王 颖, 郭艳丽, 袁洪林, 魏永锋, 闫宏涛, 陈慧慧. 光谱学与光谱分析, 2012, 32(1): 223-228
3 ZHANG XinYing, ZHENG LingNa, WANG HaiLong, SHI JunWen, FENG WeiYue, LI Liang, WANG Meng. Chinese J. Anal. Chem., 2016, (11): 1646-1651
张欣颖, 郑令娜, 王海龙, 史俊稳, 丰伟悦, 李 亮, 王 萌. 分析化学, 2016, (11): 1646-1651
4 Pozebon D, Scheffler G L, Dressler V L, Nunes M A G. J. Anal. At. Spectrom., 2014, 29(12): 2204-2228
5 Lum T S, Tsoi Y K, YueP YK, LeungK S Y. Microchem. J., 2016, 127: 94-101
6 MorenoGordaliza E, Giesen C, Lazaro A, EstebanFernandez D, Humanes B, Canas B, Panne U, Tejedor A, Jakubowski N, GomezGomez M M. Anal. Chem., 2011, 83(20): 7933-7940
7 Austin C, Fryer F, Lear J, Bishop D, Hare D, Rawling T, Kirkup L, McDonagh A, Doble P. J. Anal. At. Spectrom., 2011, 26(7): 1494-1501
8 Pozebon D, Dressler V L, Mesko M F, Matusch A, Becker J S. J. Anal. At. Spectrom., 2010, 25(11): 1739-1744
9 Niehaus R, Sperling M, Karst U. J. Anal. At. Spectrom., 2015, 30(10): 2056-2065
10 Loewen M W, KentA J R. J. Anal. At. Spectrom., 2012, 27(9): 1502-1508
11 Zhang S, He M, Yin Z, Zhu E, Hang W, Huang B. J. Anal. At. Spectrom., 2016, 31(2): 358-382
12 Horn I, Günther D. Appl. Surf. Sci., 2003, 207(14): 144-157
13 Horn I, Guillong M, Gunther D. Appl. Surf. Sci., 2001, 182(12): 91-102
14 Gunther D, Jackson S E, Longerich H P. Spectrochim. Acta B, 1999, 54(34): 381-409
15 Kuhn H R, Günther D. J. Anal. At. Spectrom., 2004, 19(9): 1158-1164
16 Benesova I, Dlabkova K, Zelenak F, Vaculovic T, Kanicky V, Preisler J. Anal. Chem., 2016, 88(5): 2576-2582
17 Paltauf G, SchmidtKloiber H. Laser.Surg. Med., 1995, 16(3): 277-287
18 WANG Qi, ZHANG Wen, WANG LiYun, LIU YongSheng, HU ShengHong, HU ZhaoChu. Spectrosc. and Spect. Anal., 2011, 31(12): 3379-3383
汪 奇, 张 文, 王立云, 刘勇胜, 胡圣虹, 胡兆初. 光谱学与光谱分析, 2011, 31(12): 3379-3383
19 Ebdon L, Foulkes M, Sutton K. J. Anal. At. Spectrom., 1997, 12(2): 213-229
20 WU ShiTou, WANG YaPing, XU ChunXue, YUAN JiHai. Chinese J. Anal. Chem., 2016, 44(7): 1035-1041
吳石头, 王亚平, 许春雪, 袁继海. 分析化学, 2016, 44(7): 1035-1041
Abstract The ablated aerosols of biological matrix sample were studied using 213 nm nanosecond laser ablation system. The stable signal intensity and high sensitivity were obtained when the laser energy was 25%, the spot size was 200 μm, the scan rate was 20 μm/s, the frequency was 20 Hz and the carrier gas was 700 mL He + 700 mL Ar. Relative fractionation index of 56 elements were investigated and 31P as the internal standard element was selected under the optimized laser ablation conditions. The results showed that particle size of the biological sample was 3 μm, which was larger compared with NIST 610 sample. Element fractionation in biological sample was smaller than in glass sample, and relative fractionation index of most elements attained 1.0 ± 0.1. Element fractionation mechanism of biological sample was discussed. The possible reason why the relative fractionation index in biological sample with large particle size did not significantly increase compared to the glass sample is that the 3μm particles entered into ICP can be atomized. On the other hand, enrichment effect for large ablation particles was relatively small. Further study of the influence factors of fractionation effect indicated that, the fractionation effect had relations with laser ablation energy, laser frequency and scan rate, negatively relation with the oxide boiling point, and positively relation with oxide bond energy and ionization energy.
Keywords Laser ablationinductively coupled plasmamass spectrometry; Nanosecond laser; Fractionation effect; Biological sample
