基于miRNA-靶位点配对的序列特征研究
2017-06-07滕少华夏飞迪刘冬宁邹小勇
滕少华,夏飞迪,张 巍,刘冬宁,王 洋,邹小勇*
(1.广东工业大学 计算机学院,广东 广州 510006;2.中山大学 化学学院,广东 广州 510275)
基于miRNA-靶位点配对的序列特征研究
滕少华1,夏飞迪1,张 巍1,刘冬宁1,王 洋2,邹小勇2*
(1.广东工业大学 计算机学院,广东 广州 510006;2.中山大学 化学学院,广东 广州 510275)
miRNA与其靶基因的作用机制十分复杂,因此,miRNA靶基因识别问题一直是miRNA研究领域的热点难题。该文基于CLASH数据集,提出了miRNA-靶位点配对序列特征,并使用随机森林建模。实验结果表明,本模型的Acc,Sen,Spe,Pre以及Mcc分别达到90.05%,89.47%,90.56%,90.43%和0.799 8;ROC和PRC的AUC分别为0.954,0.958。相比已有方法,该方法表现出更加良好的性能,说明新引入的miRNA-靶位点配对序列特征对miRNA靶基因识别有很重大的影响。
miRNA;靶基因识别;CLASH;序列分析
MicroRNAs(miRNAs)是一类内源性、长约23个核苷酸(nt)的非编码RNA,主要通过与mRNA 的3'UTR序列实现完全或不完全碱基互补配对,从而达到裂解mRNA和抑制mRNA翻译成蛋白质的目的,在后转录时期和翻译等级发挥重要的基因调节作用[1]。迄今已发现了2 000多个人类miRNA[2],这些miRNA可能调控着人体80%的基因,在各种生命活动和疾病调控中起着非常关键的作用[3]。由于miRNA靶基因识别的具体机制尚不明确,有效识别miRNA靶基因成为miRNA研究领域的一大难题。单纯用蛋白免疫印迹法[4]以及微阵列法等[5]生物实验方法来鉴别miRNA靶基因,费时而且耗费大。因此借助化学生物信息方法,挖掘miRNA潜在的靶基因,能进一步探讨miRNA作用机制和miRNA基因调控网络,具有重要理论意义和实用价值。
近十年来,研究者提出了多种生物计算方法识别miRNA靶基因。Enright 等[6]通过给miRNA与其靶基因的配对情况进行打分,计算miRNA与靶基因形成双链后的最小自由能,同时引入靶位点保守性作为最后一个条件,经过层层筛选,得到潜在的miRNA靶基因。Lewis等[7]提出了“种子”区(miRNA 5'端开始第 2到第 8位核苷酸的区间)的概念,发现种子区域的匹配情况对miRNA靶基因的识别有重大影响。Kertesz等[8]考虑了靶基因的二级结构,提出靶位点可接性概念,认为miRNA与靶基因的结合能力会受到不同二级结构影响。作为第一代生物计算方法,尽管研究人员发现了较多有用的特征,但研究表明[9-10],这些特征并不完全适用于miRNA与靶基因结合的情况。将这些特征作为筛选条件,会大大提高假阴性的预测率,于是基于机器学习的第二代生物计算的方法应运而生。
用机器学习的方法预测miRNA靶基因,其基本原理是采用可靠的数据集,根据所提出的特征,将miRNA和靶基因的结合序列特征数字化,然后将这些特征加以融合对所构建的模型进行训练,并对靶基因进行预测。Huang等[11]从表达图谱数据中提取样本用于训练模型,文献[12-14]使用交联和免疫沉淀技术(CLIP)数据用于模型训练。最近,Helwak等[15]提出的CLASH(crosslinking ligation and sequencing of hybrids)直接提供了miRNA和其对应的靶位点序列数据,为进一步研究miRNA与其靶基因位点序列作用提供了良好的平台。近年来,许多报道采用miRNA与靶位点形成双链的最小自由能,miRNA种子区域的配对数目,靶位点保守性,以及靶位点的可接入性等常用的特征[16-20]进行研究,但这些方法均有特异性太低的缺点。因此构建miRNA与靶基因结合特征,对miRNA靶基因识别具有重要意义。
本文基于CLASH实验数据集,提出了基于miRNA-靶位点配对的特征,结合一系列传统特征,运用随机森林算法建立模型,进行了miRNA靶基因识别,并与文献报道的其它两个采用同样数据集建立的模型进行了比较。
1 实验部分
1.1 数据提取与处理
在CLASH实验中提取了18 514条数据,其中包含399个miRNAs和18 514个靶位点数据,并将这些样本数据全部作为正样本,每一条样本中均包括miRNA和靶位点信息。由于负样本匮乏,将CLASH数据集中所有的miRNAs和靶位点进行一一配对,共有3 693 543个数据组合。然后去除其中的正样本,按正负样本1∶1比例,从3 675 029条数据中随机选择18 514条数据作为本研究所需的负样本。为了研究随机样本对所构建模型的影响,进行了多次重复实验来检测模型的鲁棒性。
1.2 特征集合
共选用26种特征(84个特征值),具体的特征集合如表1所示。其中前21种特征共57个特征值,文献[16-20]已有报道;本文所构建的后5种特征(ID 22~26),含有27个特征值,这些特征值充分考虑了miRNA与其靶基因的作用情况。

表1 miRNA与靶位点结合特征集合Table 1 Feature sets of the interaction of miRNA and target sites
1.3 特征选择
特征选择针对高维度数据计算问题而提出,通过剔除冗余特征和无关特征,提高机器学习算法的泛化性能和运行效率。本文使用最小冗余最大相关算法(Minimal redundancy maximal relevance criterion,mRMR)[21]对84个特征排序,并选择了最优特征子集构建模型。
1.4 随机森林
随机森林[22]是一种组合方法,由许多的决策树组成,因这些决策树的形成采用了随机的方法,因此也称作随机决策树。随机森林中的树之间没有关联,当测试数据进入随机森林时,让每一棵决策树进行分类,最后取所有决策树中分类结果最多的那类为最终结果。本文使用随机森林机器学习方法作为训练模型,算法来源于Scikit-learn(http://scikit-learn.org/stable/)工具包[23],整个程序使用python开发,并优化了森林中树的数目和每棵树的特征数两个参数。
1.5 性能指标
分类器的性能可通过一些独立的指标进行评估。为评估模型的性能,以准确率(Acc)、敏感度(Sen)、特异性(Spe)、精确度(Pre)和马氏相关系数(Mcc) 5种指标来评估模型的性能。这些指标的计算方法如下:

其中,TP指被判定为正样本,也是正样本的数目;TN指被判定为负样本,也是负样本的数目;FN指被判定为负样本,但事实上是正样本的数目;FP指被判定为正样本,但事实上是负样本的数目。此外,受试者工作特征曲线(Receiver operating characteristic curve,ROC曲线)和 准确率-召回率曲线(Precision-recall curve,PRC曲线)也用于评估模型的性能。ROC曲线是反映敏感性和特异性连续变量的综合指标,采用构图法揭示敏感性和特异性的相互关系,将连续变量设定出多个不同的临界值,从而计算出一系列敏感性和特异性。再以敏感性为纵坐标、(1-特异性)为横坐标绘制成曲线,曲线下面积越接近1,表明模型性能越好。PRC曲线是反映准确率和召回率(敏感性)连续变量的综合指标,采用构图法揭示准确率和敏感性的相互关系,将连续变量设定出多个不同的临界值,从而计算出一系列准确率和敏感性。再以准确率为纵坐标、敏感性为横坐标绘制成曲线,曲线下面积越接近1,表明模型性能越好。
1.6 实验流程
miRNA的靶基因预测属于机器学习问题,整个实验的流程如图1所示。
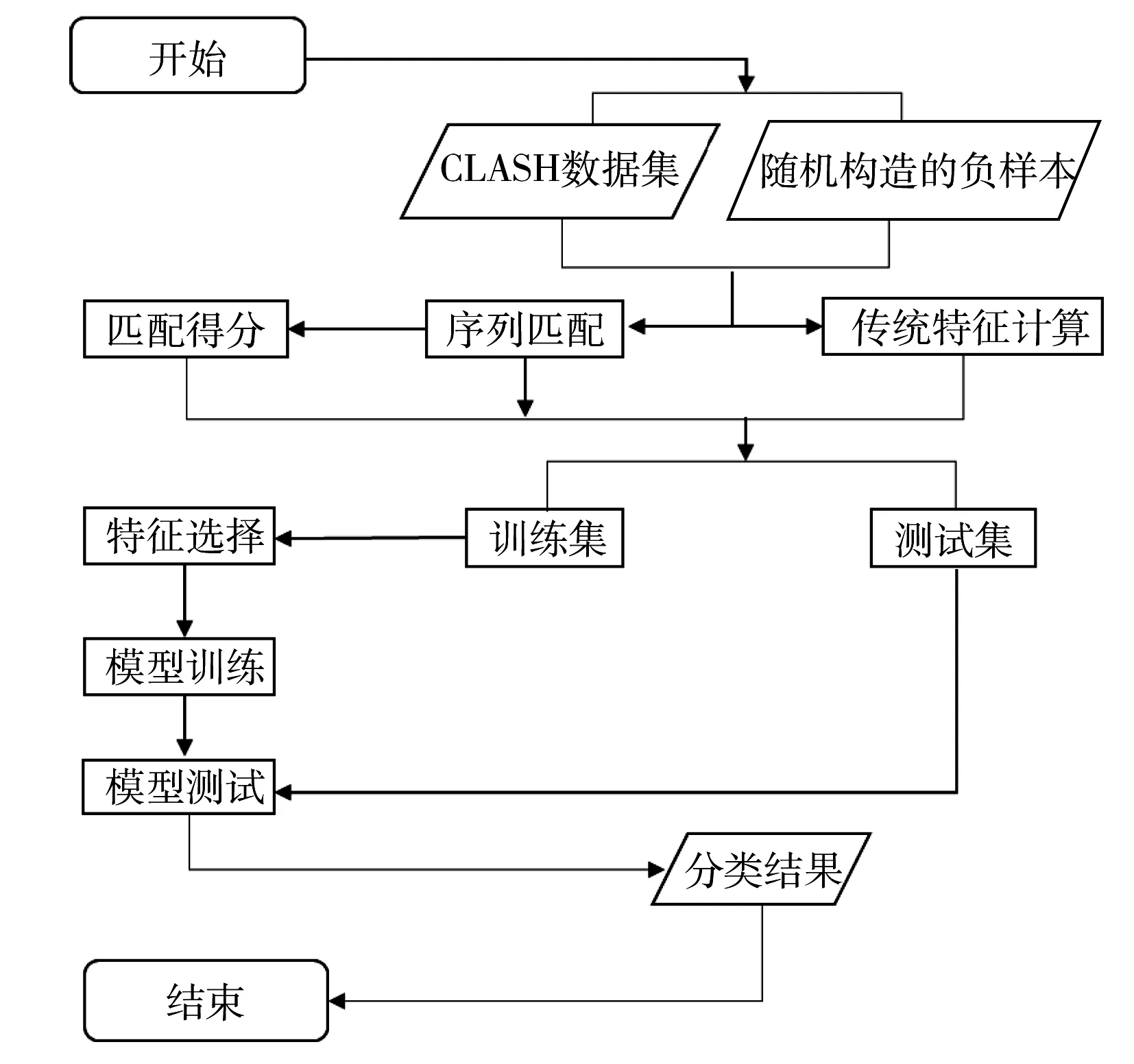
图1 实验流程图Fig.1 The flow chart of the experiment
步骤1:选择CLASH数据集作为正样本,并根据该数据集构造负样本,将CLASH数据集中的miRNA与靶位点序列随机配对,删除其中的正样本,再从剩余的数据集中随机选择18 514条作为负样本,正负样本比例为1∶1;
步骤2:根据传统特征的计算方法,计算样本传统特征的特征值;
步骤3:采用改进的Smith-Waterman方法将正负样本进行序列匹配,并转换为二进制序列。再根据正样本序列匹配的情况构造权重向量W,并以此向量计算正负样本的序列匹配得分特征;
步骤4:采用随机森林的方法构建模型,并训练模型的参数;

图2 序列匹配二进制化表示Fig.2 A binary representation of sequence match

图3 正负样本匹配对比Fig.3 Comparison of positive and negative samples matching
步骤5:模型测试;
步骤6:与其他模型比较并分析。
2 结果与讨论
2.1 基于miRNA-靶位点的配对miRNA与其靶位点并不完全匹配,且匹配情况差异很大。本文根据样本集中miRNA与其靶位点的配对情况,将每一条miRNA与靶位点结合后的双链表示为由“0”和“1”组成的二进制序列,并对构成的二进制序列进行分析,具体过程如图2所示,其中阴影部分为“种子区域”。
在图2中,BEYLA序列为miR-149对应的靶位点序列。首先采用改进的Smith-Waterman方法[24],按照碱基A∶U和G∶C互补配对原则进行序列匹配,允许G∶U错配。从miR-149序列5'端的第1个核苷酸开始,与BEYLA序列的每1个核苷酸进行比对,如果匹配,则用“1”表示,对应的核苷酸在其相应的位置用竖线“|”连接;如果不匹配,则用“0”表示。每1条序列中均可能有一些短横线“-”,表示该位置不含任何核苷酸。因此,miR-149序列和BEYLA靶位点序列的匹配可转化为二进制序列“11111111011110111110010”,共含有23个“0”和“1”特征值。因为CLASH数据集中大部分miRNA的长度为23,因此本文将每一条miRNA与靶位点结合后的双链转换为23个“0”或“1”组成的特征值序列,如果miRNA的长度小于23,则该特征值用0补充,如果miRNA长度大于23,多出来的特征值不予考虑。最后,本文将这23个特征值加入特征集。
采用以上数字编码的方法,对CLASH数据集和随机构造的负样本进行了比对分析。首先,将每一个样本进行了序列匹配,然后转换为二进制“0”和“1”序列,并统计每个位置配对成功的概率,结果如图3所示。
在图3中,横轴表示miRNA每个核苷酸的位置,纵轴表示miRNA上每个位置配对成功的概率。图中上面的曲线表示正样本中miRNA上每个位置配对成功的概率,下面的曲线表示负样本中miRNA上每个位置配对成功的概率。从图3可以发现,正样本整体的匹配情况比负样本好,特别是第20位之前的匹配情况,正样本明显优于负样本。同时实验还发现,正负样本序列两端配对成功的概率远远低于中间核苷酸位置配对成功的概率。本文对每一个位置的差异值进行计算,结果发现,正负样本在第2到第8位的配对情况的差异相对于其他位置大很多,这与之前的研究观点一致[25],即miRNA种子区域的配对情况对miRNA的靶基因识别具有很重要的作用。
基于上述发现,本文根据正样本中每个位置的匹配成功率构造了一个权重向量w,并以此向量为基础,提出了几种方法对miRNA的匹配序列进行打分,得到了4个关键特征。
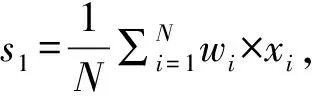
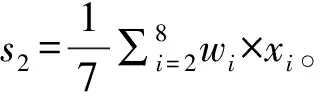
“全序列匹配特征1” 和“种子区域匹配特征1”考虑了配对成功对miRNA靶基因识别的影响。
特征3:对于miRNA上第i位的匹配情况xi=1,如果xi=1,其对应的权值为wi;如果x1=0,其对应的权值则为qi=1-wi,构建了“全序列匹配特征2”,可以通过计算整段序列匹配得分的平均值s3,其计算公式如下,其中N(N=23)为序列长度。
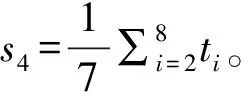
其中,特征3和特征4,既考虑了匹配成功情况,也考虑了匹配不成功的情况。因此,根据miRNA-靶位点配对情况,构建了23个序列特征和“全序列匹配特征1”、“种子区域匹配特征1”、“全序列匹配特征2” 和“种子区域匹配特征2”的4个序列得分特征,共27个特征值。
2.2 特征选择
根据表1构建了包含84个特征的特征集,为了研究各特征的贡献,采用mRMR方法对各特征进行排序,前29个特征排名如表2所示。由表2可见,所构建的“种子区域匹配特征1”排名第4,“全序列匹配特征1”排名第5,“种子区序列匹配特征2”排名第8,“全局序列匹配特征2”排名第9。说明新构建的特征对miRNA靶基因识别具有相当重要的作用。同时还可以看到,传统特征如最小自由能、保守性和种子区域配对均对miRNA靶基因识别起着重要作用。

表2 前29个特征排名Table 2 The top 29 features sorted by mRMR

图4 基于不同特征子集的预测结果Fig.4 The prediction results based on the various feature subset
根据各特征的排名,以1为梯度,分别使用排名前84,83,…,3,2,1个特征组成的特征子集,然后基于每一个特征子集构建对应的模型,计算了Acc,Sen,Spe,Pre以及Mcc,以考察所构建模型的性能,具体结果如图4所示。从图4可以看出,当特征子集中的特征数大于29时,模型性能基本无变化,因此本文最终选择前29个特征作为特征子集。在排名前29个特征中,本文提出的特征共有13个(如表2的阴影所示),表明本文提出的特征是可行的。
2.3 参数训练
随机森林有两个重要的参数,n_estimators表示森林中树的棵数,max_feature表示每次生成决策树时选择的特征个数。对于n_estimators,以100梯度,提取100~1 000的所有取值(100,200,…,1 000)。对于max_feature,研究了scikitlearn软件包中所有取值。结果表明,当n_estimators=400和max_feature=4时,模型的性能达到最佳。
2.4 鲁棒性评估
依据上述步骤,建立了基于随机森林算法的模型,对miRNA靶基因进行预测。为研究模型的鲁棒性,将负样本进行了10次随机采样,根据所建立的数据集,构建模型和计算各性能指标,具体结果如表3所示。
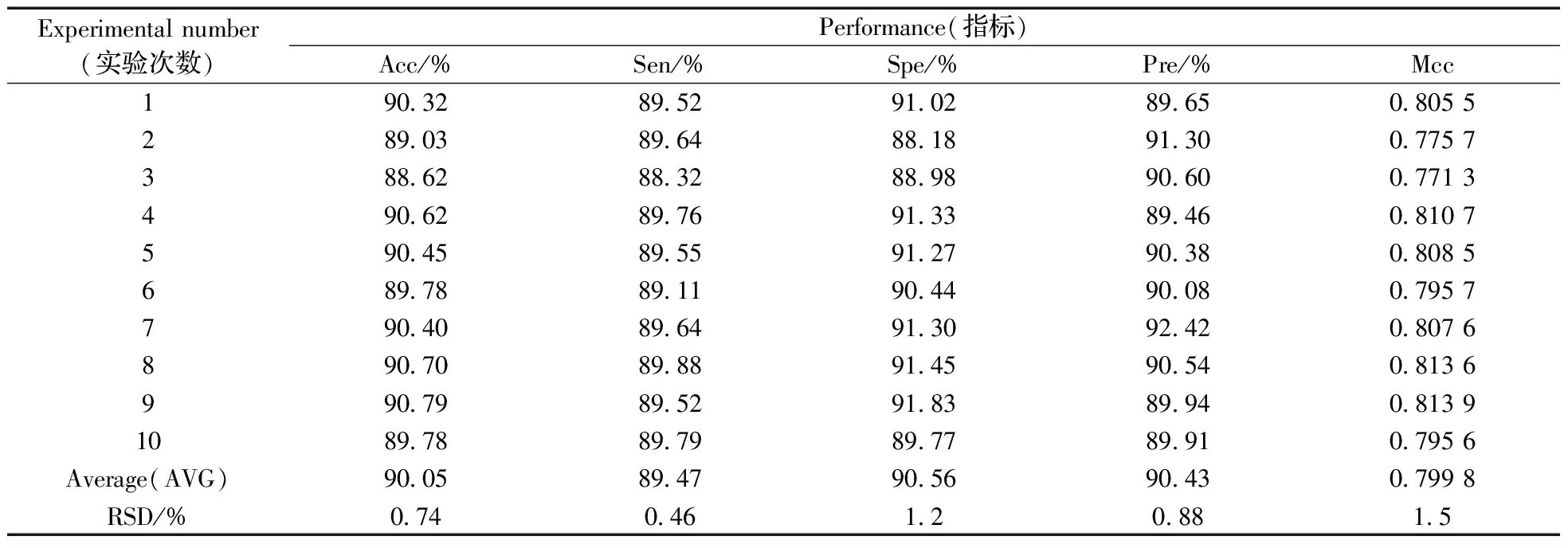
表3 模型鲁棒性评估的结果Table 3 The results of the robustness of the model
从表3可以看出,准确率(Acc)、敏感度(Sen)、特异性(Spe)、精确度(Pre)、马氏相关系数(Mcc)的平均值分别为90.05%,89.47%,90.56%,90.43%,0.799 8,而且RSD均小于1.6%。结果表明,本文所建立的模型具有很强的鲁棒性。同时,基于最高的Acc值,本文还计算了模型的ROC和PRC值。结果显示,该模型的ROC和PRC曲线下面积分别可达到0.953 7,0.958 4,说明模型对于靶基因预测表现出良好的性能。
2.5 与其他方法比较
为了验证新构建特征的有效性,基于传统特征集构建了miRNA靶基因预测模型,并与本文所使用的模型进行比较。结果表明,当加入新构建的特征后,模型的性能得到了很大的提高,Acc提高了6%,Spe和Pre均提高了近5%,Sen的改善明显(提高了近9%),ROC和PRC的AUC提高了10%左右,进一步验证了新构建特征的有效性。同时,将本方法与已有的MirTarget[26]和TarPmiR[27]方法进行比较(表4),可以看到,本文所采用的模型整体性能更好。其中本方法的Acc相比TarPmiR和MirTarget分别增加了8%和5%,实验结果明显改善。同时本模型的ROC和PRC的AUC高达0.95以上,也验证了本模型性能的稳定性。
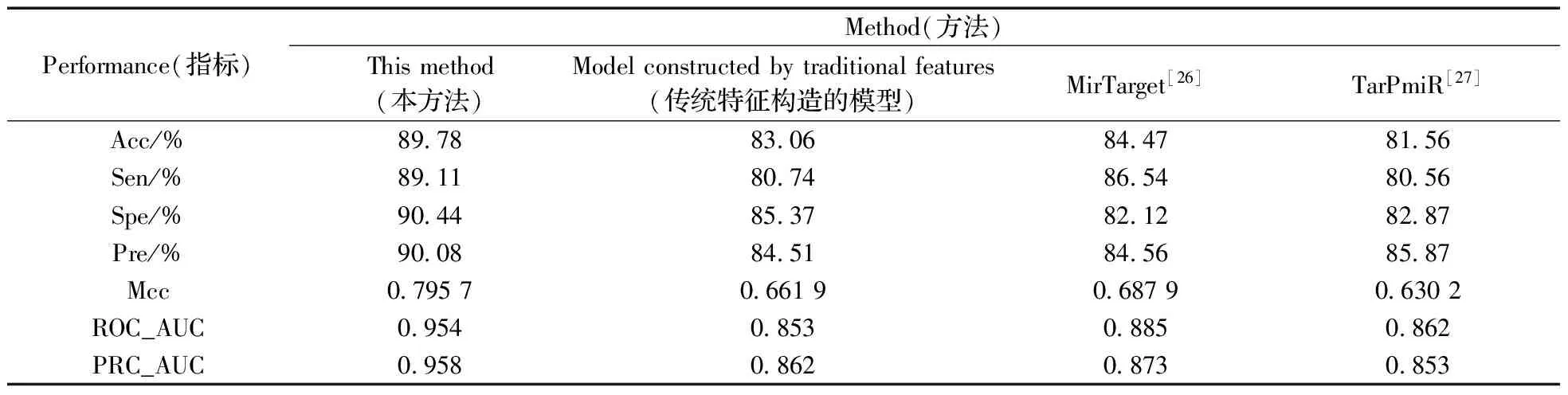
表4 不同方法的比较Table 4 The comparision of different method
3 结 论
本文基于CLASH数据集,构造了27个miRNA-靶位点配对序列相关特征,结合传统特征,组成了1个包含84个特征值的特征集合,并使用随机森林模型进行机器学习,构造miRNA靶基因预测模型。本模型的平均Acc,Sen,Spe,Pre以及Mcc分别达到90.05%,89.47%,90.56%,90.43%和0.799 8,ROC和PRC的AUC分别为0.954和0.958,表现出良好的预测性能。根据实验结果可以预见,所建立的方法可为miRNA靶基因预测提供一定的技术支撑。
[1] Bartel D P.Cell,2004,116(2):281-297.
[2] Kozomara A,Griffithsjones S.NucleicAcidsRes.,2014,42(D1):68-73.
[3] Ambros V.Nature,2004,431(7006):350-355.
[4] Lagos Q M,Rauhut R,Lendeckel W,Tuschl T.Science,2001,294(5543):853-858.
[5] Lin L P,Lau N C,Garrett-Engele P,Grimson A,Schelter J M,Castle J,Bartec D P,Linsley P S,Johnson J M.Nature,2005,433(7027):769-773.
[6] Enright A J,John B,Gaul U,Tuschl T,Sander C.GenomeBiol.,2003,5(1):R1.
[7] Lewis B P,Shih I H,Jones-Rhoades M W,Bartel D P,Burge C B.Cell,2004,115(7):787-798.
[8] Kertesz M,Iovino N,Unnerstall U,Gaul U,Segal E.NatureGenetics,2007,39(10):1278-1284.
[9] Didiano D,Hobert O.Nat.Struct.Mol.Biol.,2006,13(9):849-851.
[10] Sethupathy P,Megraw M,Hatzigeorgiou A G.Nat.Methods,2006,3(11):881-886.
[11] Huang J C,Babak T,Corson T W,Chua G,Khan S,Gallie B L,Hughes T R,Blencowe B J,Frey B J,Morris Q D.Nat.Methods,2007,4(12):1045-1049.
[12] Liu C,Mallick B,Long D,Rennie W A,Wolenc A,Carmack C S,Ding Y.NucleicAcidsRes.,2013,41(14):338-345.
[13] Reczko M,Maragkakis M,Alexiou P,Grosse I,Hatzigeorgiou A G.Bioinformatics,2012,28(6):771-776.
[14] Gumienny R,Zavolan M.NucleicAcidsRes.,2015,43(3):1380-1391.
[15] Helwak A,Kudla G,Dudnakova T,Tollervey D.Cell,2013,153(3):654-665.
[16] Yousef M,Jung S,Kossenkov A V,Showe L C,Showe M K.Bioinformatics,2007,23(22):2987-2992.
[17] Wang X.Bioinformatics,2014,30(10):1377-1383.
[18] Shuang C,Guo M,Wang C,Wang C,Liu C Y,Liu Y.IEEE/ACMTrans.Comput.Biol.Bioinf.,2015,13(6):1145-1169.
[19] Yu S,Kim J,Min H,Yoon S.Methods,2014,69(3):220-229.
[20] Bandyopadhyay S,Mitra R.Bioinformatics,2009,25(20):2625-2631.
[21] Ponsa D,López A.IberianConferenceon-PatternRecognitionandImageAnalysis,2007,4477:47-54.
[22] Liaw A,Wiener M.RNews,2001,2(3):18-22.
[23] Pedregosa F,Varoquaux G,Gramfort A,Michel V,Thirion B,Grisel O,Blondel M,Prettenhofer P,Weiss R,Dubourg V,Vanderplas J,Passos A,Cournapeau D,Brucher M,Perrot M,Duchesnay E.J.Mach.Learn.Res.,2013,12(10):2825-2830.
[24] Smith T F,Waterman M S,Fitch W M.J.Mol.Evol.,1981,18(1):38-46.
[25] Agarwal V,Bell G W,Nam J W,Bartel D P.Elife,2015,4:e05005.
[26] Wang X.Bioinformatics,2016,32(9):1316-1322.
[27] Ding J,Li X M,Hu H Y.Bioinformatics,2016,32(18):2768-2775.
Study of Sequence Features Based on MicroRNA-Target Sites Pairing
TENG Shao-hua1,XIA Fei-di1,ZHANG Wei1,LIU Dong-ning1,WANG Yang2,ZOU Xiao-yong2*
(1.Faculty of Computer,Guangdong University of Technology,Guangzhou 510006,China;2.School of Chemistry, Sun Yat-sen University,Guangzhou 510275,China)
The mechanisms underlying the interaction of miRNAs with their mRNA targets are quite complex,which makes miRNA target prediction be a hot issue in the field of miRNA research.The features of miRNA-target sites pairing sequences were proposed based on the CLASH dataset,and the random forest models were applied for modeling.The average values of Acc,Sen,Spe,Pre and Mcc are 90.05%,89.47%,90.56%,90.43% and 0.799 8,respectively,and the AUC of ROC and PRC are 0.954 and 0.958,respectively.The results indicated that the current method shows a better performance compared with the existed methods, and the features newly constructed have a very significant impact on the identification of miRNA target genes.
miRNA;target prediction;CLASH;sequence analysis
2016-12-12;
2017-01-20
国家自然科学基金项目(21675180);广东省科技计划项目(2014A040401022,2015A030401033,2016B010108007);广州市科技计划项目(201604020145)
10.3969/j.issn.1004-4957.2017.05.006
O629.74;TB9
A
1004-4957(2017)05-0614-07
*通讯作者:邹小勇,博士,教授,研究方向:化学计量学、电分析化学,Tel:020-84114919,E-mail:ceszxy@mail.sysu.edu.cn
