病毒感染宿主细胞可能性的序列非比对法评估*
2017-05-30刘雪梅臧翔黄天来杨哲李文叶宇中胡珊
刘雪梅 臧翔 黄天来 杨哲,2 李文 叶宇中 胡珊
(1.华南理工大学 物理与光电学院, 广东 广州 510640; 2.中国工商银行 广州东城支行, 广东 广州 510100;3.中山大学 中山医学院计算机中心, 广东 广州 510275)
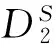
1 序列非比对法原理
(1)
式中:CSi(W)为在序列Si中W出现的次数,W为序列中所有可能的k-tuple组成的集合;wi为k-tuple元素;A为字母表,是一组符号或字符,字母表中的元素组成序列.
(2)
式中,pW为W在序列中出现的概率.
(3)
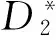
2 病毒和宿主细胞的DNA序列非比对比较
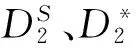
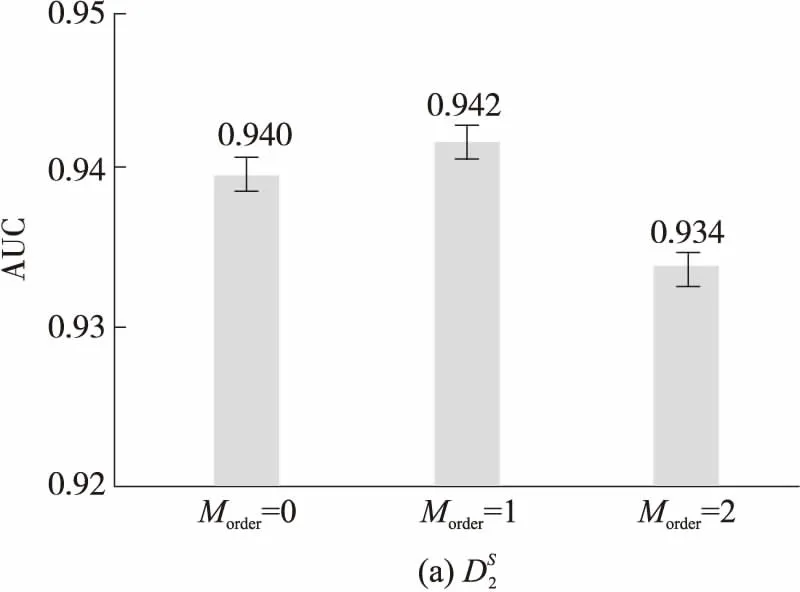
图1 k=5、Morder=1,2,3时和的AUC柱状图
2.2 最佳阈值的确定
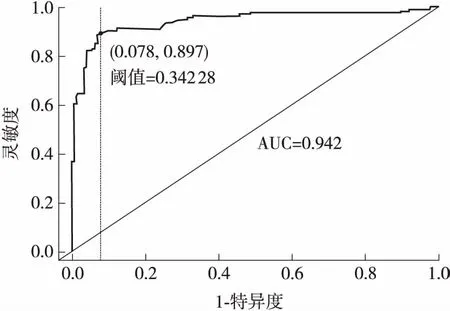
图2 k=5、Morder=1时的ROC曲线
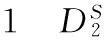
表和统计打分值
2.3 最佳阈值应用实例
3 结语
:
[1] WOMMACK K E,COLWELL R R.Virioplankton:viruses in aquatic ecosystems [J].Microbiol Mol Biol Rev,2000,64(1):69- 114.
[2] WEINBAUER M G.Ecology of prokaryotic viruses [J].FEMS Microbiol Rev,2004,28(2):127- 181.
[3] SUTTLE C A.Marine viruses-major players in the global ecosystem [J].Nat Rev Microbiol,2007,5(10):801- 812.
[4] CRAM J A,LI C X,NEEDHAM D M,et al.Cross-depth analysis of marine bacterial networks suggests downward propagation of temporal changes [J].ISME Journal,2015,9(12):2573- 2586.
[5] QI J,WANG B,HAO B I.Whole proteome prokaryote phylogeny without sequence alignment:aK-string composition approach [J].Journal of Molecular Evolution,2004,58(1):1- 11.
[6] DOMAZETLOSO M,HAUBOLD B.Alignment- free detection of local similarity among viral and bacterial genomes [J].Bioinformatics,2011,27(11):1466- 1472.
[7] REINERT G,CHEW D,SUN F,et al.Alignment-free sequence comparison(I):statistics and power [J].Journal of Computational Biology:A Journal of Computational Molecular Cell Biology,2009,16(12):1615- 1634.
[8] LIPPERT R A,HUANG H,WATERMAN M S.Distributional regimes for the number ofk-word matches between two random sequences [J].Proceedings of the National Academy of Sciences of the United States of America,2002,99(22):13980- 13989.
[9] WANG Y,LEI X,WANG S,et al.Effect ofk-tuple length on sample-comparison with high-through put sequencing data [J].Biochemical & Biophysical Research Communications,2015,469(4):1021- 1027.
[10] SONG K,REN J,LIU X M,et al.Alignment-free sequence comparison based on next-generation sequencing reads [J].Journal of Computational Biology,2012,20(2):64- 79.
[11] ZHAI Z,KU S Y,LUAN Y,et al.The power of detecting enriched patterns:an HMM approach [J].Journal of Computational Biology:A Journal of Computational Molecular Cell Biology,2010,17(4):581- 592.
[12] WANG Y,LIU L,CHEN L,et al.Comparison of meta-transcriptomic samples based onk-tuple frequencies [J].PLoS ONE,2014,9(1):e84348/1- 19.
[13] 刘雪梅,文德华,於黄忠,等.基于D2shepp统计法的非序列局部比对 [J].华南理工大学学报(自然科学版),2012,40(8):106- 110.
LIU Xue-mei,WEN De-hua,YU Huang-zhong,et al.Local alignment-free sequences based on D2shepp statistics [J].Journal of South China University of Technology(Natural Science Edition),2012,40(8):106- 110.
[14] LIU X M,WAN L,LI J,et al.New powerful statistics for alignment-free sequence comparison under a pattern transfer model [J].Journal of Theoretical Biology,2011,284(1):106- 116.
[15] WAN L,REINERT G,SUN F,et al.Alignment-free sequence comparison(II):theoretical power of comparison statistics [J].Journal of Computational Biology,2010,17(11):1467- 1490.
[16] BAI J,KAI S,JIE R,et al.Comparison of metagenomics samples using sequence signatures [J].BMC Genomics,2012,13(1):138- 140.
