基于高通量测序的鄱阳湖典型湿地土壤细菌群落特征分析
2017-05-25王鹏,陈波,张华
王 鹏,陈 波,张 华
1 江西师范大学鄱阳湖湿地与流域研究教育部重点实验室, 南昌 330022 2 江西师范大学地理与环境学院, 南昌 330022
基于高通量测序的鄱阳湖典型湿地土壤细菌群落特征分析
王 鹏1,2,*,陈 波1,2,张 华1,2
1 江西师范大学鄱阳湖湿地与流域研究教育部重点实验室, 南昌 330022 2 江西师范大学地理与环境学院, 南昌 330022
采用高通量测序技术分析了鄱阳湖典型湿地土壤细菌群落特征。测序结果表明,不同植被土壤细菌群落丰度与多样性的排序相同:苔草带>苔草-虉草带>芦苇带>泥滩带>藜蒿带。沿湖面至坡地,空间位置相近的土壤细菌群落结构具有更大的相似性,苔草-虉草带、苔草带和芦苇带的细菌群落结构相近,泥滩带和藜蒿带的细菌群落结构差异较大。变形菌门(30.0%)是湿地土壤平均相对丰度最高的门,其次为酸杆菌门(16.7%)和绿弯菌门(16.5%);多数门分类细菌相对丰度沿湖面至坡地存在一定变化趋势。硝化螺菌属是第一大属分类水平细菌群落。在土壤化学指标中,与鄱阳湖湿地细菌群落相关性较大的是总磷、铵态氮和有机质含量。以上研究结果表明,鄱阳湖湿地不同植被土壤细菌群落具有结构性差异,但沿湖面至坡地存在规律性变化。
鄱阳湖湿地;高通量测序;细菌多样性;细菌群落结构
湿地作为陆地与水生生态系统的过渡区域,独特的水土界面环境和较高的生物生产力促进了碳、氮、磷等元素的快速循环转化,是陆地地表元素迁移和转化的重要场所。微生物是湿地生态系统的分解者,在湿地物质能量循环中起着重要作用[1]。查明湿地微生物群落结构及分布特征,有助于分析微生物与环境因子的关系,阐明湿地元素迁移转化的机理,为制定合理的湿地管理策略提供科学依据。随着分子生物学的发展,尤其是低成本、自动化的高通量测序技术[2]的应用,能同时对样品中的优势物种、稀有物种及一些未知的物种进行检测,获得样品中的微生物群落组成,在水体[2- 5]、沉积物[6- 7]和土壤[8- 10]微生物群落结构研究中得到广泛应用。
鄱阳湖是我国第一大淡水湖,也是生态多样性丰富的淡水湿地系统。鄱阳湖湿地是我国重要的生态功能保护区,是世界自然基金会划定的全球重要生态区,承担着调洪蓄水、调节气候、降解污染等多种生态功能,也是白鹤、东方白鹳和小天鹅等数百种珍稀水鸟的越冬地[11]。由于鄱阳湖湖盆的独特自然地理特征,鄱阳湖湿地生态环境受水位变化影响大,湿地植物群落沿水分梯度分布,呈现条带状的总体格局[12- 13]。鄱阳湖湿地不同植被土壤微生物群落结构有何特征?这种群落结构特征与碳、氮、磷等土壤化学指标有何关系?阐明这些问题对深入理解鄱阳湖湿地生态系统的物质和能量循环具有重要意义。目前鄱阳湖湿地微生物群落的研究较少,主要包括不同湿地植物土壤微生物量[14]、土壤微生物群落对碳源的利用[15]、湖泊沉积物及水体的微生物群落结构[7,16- 17]等方面的研究,尚缺少对鄱阳湖不同湿地植物土壤微生物群落结构及其与环境因子关系的研究。
本次研究选择鄱阳湖典型自然湿地,基于高通量测序分析不同湿地植被土壤的细菌群落特征,探讨细菌群落与土壤化学指标的关系,为鄱阳湖生态管理提供科学依据。
1 材料与方法
1.1 研究区概况
本次研究选择赣江主支河口附近的鄱阳湖典型湿地进行,该典型湿地(图 1)地形由陆地向湖区逐渐倾斜,走向与湖水退水方向一致。最高处毗邻赣江河堤,高程约为18.4 m。近湖区地势相对平坦,与鄱阳湖大湖面相接,高程约为11.2 m。湿地受水位季节性变化影响显著,枯水期水位下降,沿湖面至坡地分布泥滩带、苔草-虉草带、苔草带、芦苇带和藜蒿带;丰水期水位上涨,除藜蒿带和部分芦苇带外大部分洲滩被淹没。洲滩土壤粒径由湖面至坡地逐渐变大,砂性土壤比例增大。

图1 研究区位置及采样点示意图 Fig.1 Sketch map of study area and sampling sites
1.2 土样采集与理化分析
枯水期2014年11月2日分别在泥滩带、苔草-虉草带、苔草带、芦苇带和藜蒿带采集土壤样品。每处土样采用S型5点取样法,取样深度为0—10 cm。采集的每处5个土样分为两部分,一部分用于化学指标测试,另一部分充分混合用于DNA提取和高通量测序。
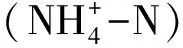
1.3 细菌16S rRNA 基因测序
采用E.Z.N.A.® Soil DNA Kit(Omega Bio-tek, Norcross, GA, U.S.)提取土壤总DNA,利用1%琼脂糖凝胶电泳检测抽提的基因组DNA。对16S rRNA基因的V3-V4高变区片段进行PCR扩增,引物序列[18]为338F(5′- ACTCCTACGGGAGGCAGCA- 3′)和 806R(5′- GGACTACHVGGGTWTCTAAT- 3′)。扩增条件为:95 °C预变性2 min,接着进行25个循环,包括95°C变性30 s,55°C退火30 s,72°C延伸30 s;循环结束后72°C最终延伸5 min。每个样本3个重复,将同一样本的PCR产物混合后用2%琼脂糖凝胶电泳检测,使用AxyPrepDNA凝胶回收试剂盒(AXYGEN公司)切胶回收PCR产物,Tris_HCl洗脱;2%琼脂糖电泳检测。参照电泳初步定量结果,将PCR产物用QuantiFluorTM-ST蓝色荧光定量系统(Promega公司)进行检测定量,按照每个样本的测序量要求,进行相应比例的混合。测序在上海美吉生物医药科技有限公司的Illumina Miseq PE300平台进行。
1.4 生物信息处理
利用Mothur(V.1.36.1)对原始DNA序列进行过滤处理,去除嵌合体,得到优化序列;按照97%相似性将优化序列划分可操作分类单元(OTU,Operational Taxonomic Units);基于OTU进行稀释性曲线分析,并计算Chao1丰度指数、覆盖度(Coverage)和Shannon多样性指数。利用主成分分析(PCA)分析各样间OTU相似性。对比Silva(Release119, http://www.arb-silva.de)16S rRNA数据库,采用RDP Classifier(http://rdp.cme.msu.edu/)贝叶斯算法对97%相似水平的OTU代表序列进行分类学分析,并在各个分类水平上统计每个样品的群落组成;利用冗余分析研究土壤化学指标与细菌群落的关系。
2 结果
2.1 细菌群落丰度、多样性和结构差异
5个土壤样品的高通量测序共得到134 223条高质量序列,平均长度为438.50 bp。以97%相似度划分,共得到2 072个OTUs。表 1为5个土壤样品中的优化序列、OTU数量及多样性指数。各样品文库的覆盖率(Coverage)范围为99.04%—99.65%,说明土样中基因序列被检出的概率很高,本次测序结果能够代表湿地土壤细菌群落的真实情况。
表 1为反映鄱阳湖湿地土壤细菌丰度和多样性的相关指数。Chao1指数用于估算样品中所含OTU总数,反映了菌群丰度,Chao1越大说明细菌群落丰度越高。Shannon指数反映细菌群落alpha多样性指数,Shannon值越大,说明细菌群落多样性越高。由表1可知,5种植被土壤细菌群落丰度与多样性的排序是一致的:苔草带(S3)>苔草-虉草带(S2)>芦苇带(S4)>泥滩带(S1)>藜蒿带(S5)。
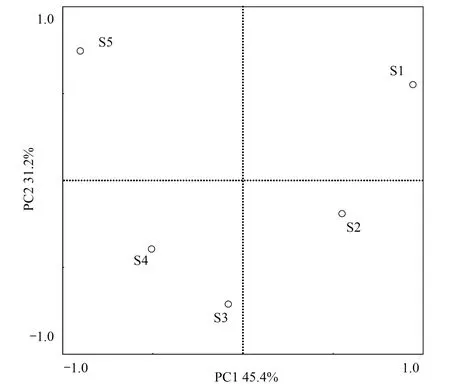
图2 采样点土壤OTU的主成分分析 Fig.2 Principal component analysis of OTU in sampling soils
对5种土壤中OTU的组成进行PCA分析(图 2),第一主轴和第二主轴的贡献率分别为45.4%和31.2%。苔草带(S3)和芦苇带(S4)之间的距离最近,表明两者土壤OTU组成相近;苔草-虉草带(S2)和泥滩带(S1),以及苔草-虉草带(S2)和苔草带(S3)的土壤OTU组成也相近,但差异性大于苔草带(S3)和芦苇带(S4);藜蒿带(S5)和其它4种植被土壤OTU组成的差异都较大,其中与芦苇带(S4)的差异相对较小。沿湖面至坡地,空间位置相近的土壤细菌群落结构也相近;苔草-虉草带(S2)、苔草带(S3)和芦苇带(S4)的细菌群落结构相对相近,泥滩带(S1)和藜蒿带(S5)的细菌群落结构差异较大。
2.2 细菌门水平分类和属水平分类
5个鄱阳湖湿地土壤样品的2 072条OTUs分属于37个门,86个纲,176个目,294个科,401个属,718个种。图 3为门水平上的细菌分类。相对丰度较高的分别为变形菌门(Proteobacteria,30.0%),酸杆菌门(Acidobacteria,16.7%),绿弯菌门(Chloroflexi,16.5%),硝化螺旋菌门(Nitrospirae,10.2%),厚壁菌门(Firmicutes,7.5%),放线菌门(Actinobacteria,4.8%),芽单胞菌门(Gemmatimonadetes,3.8%)和绿菌门(Chlorobi,1.8%)。变形菌门(Proteobacteria)是鄱阳湖湿地土壤的优势类群,主要包括α-变形菌纲(Alphaproteobacteria,7.2%)、β-变形菌纲(Betaproteobacteria,8.9%)、δ-变形菌纲(Deltaproteobacteria,10.5%)和γ-变形菌纲(Gammaproteobacteria,2.8%);ε-变形菌纲(Epsilonproteobacteria)平均丰度仅为0.1%。
苔草-虉草带(S2)、苔草带(S3)和芦苇带(S4)土壤具有相近的细菌群落结构(图 2),相对丰度最高的都是变形菌门(Proteobacteria),分别为31.9%,45.8%和35.5%。除变形菌门(Proteobacteria)外,苔草-虉草带(S2)相对丰度较高的分别为绿弯菌门(Chloroflexi,16.1%)和厚壁菌门(Firmicutes,15.3%);苔草带(S3)相对丰度较高的分别为硝化螺旋菌门(Nitrospirae,13.5%)和酸杆菌门(Acidobacteria,8.5%);芦苇带(S4)相对丰度较高的分别为酸杆菌门(Acidobacteria,18.8%)和绿弯菌门(Chloroflexi,11.6%)。与其它3种植被土壤相比,泥滩带(S1)和藜蒿带(S5)细菌群落结构差异较大(图 2);泥滩带(S1)相对丰度最高的是绿弯菌门(Chloroflexi,24.9%),其次为硝化螺旋菌门(Nitrospirae,20.2%)和变形菌门(Proteobacteria,17.6%);藜蒿带(S5)相对丰度最高的是酸杆菌门(Acidobacteria,32.0%),其次为绿弯菌门(Chloroflexi,22.4%)和变形菌门(Proteobacteria,19.1%)。
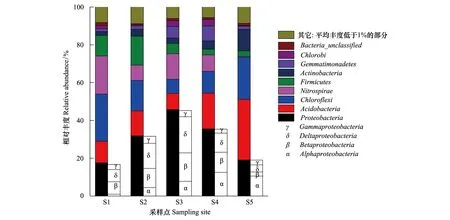
图3 鄱阳湖湿地土壤细菌门水平分类 Fig.3 Relative abundances of bacterial phyla in Poyang Lake wetland soils
沿湖面至坡地,5种植被土壤的门分类细菌相对丰度存在一定变化趋势(图 3)。变形菌门(Proteobacteria)相对丰度存在先增大,后减小的趋势,在苔草带(S3)达到最大;酸杆菌门(Acidobacteria)沿湖面至坡地相对丰度大致呈不断增大的趋势;绿弯菌门(Chloroflexi)沿湖面至坡地,存在先减小后增大的趋势,在苔草带(S3)达到最小;硝化螺旋菌门(Nitrospirae)不存在明显变化趋势,在泥滩带(S1)和苔草带(S3)相对丰度较高,在藜蒿带(S5)则很小。厚壁菌门(Firmicutes)在苔草-虉草带(S2)达到最大,沿湖面至坡地大致呈不断减小的趋势;放线菌门(Actinobacteria)沿湖面至坡地大致呈增大趋势;芽单胞菌门(Gemmatimonadetes)沿湖面至坡地呈先增大后减小趋势;绿菌门(Chlorobi)沿湖面至坡地没有明显变化趋势。
图 4为属分类水平上的细菌分类,将平均丰度低于1.5%的部分合并为others在图中显示,剩余16个属水平分类中有5个属于分类学数据库分类学谱系的中间等级,没有科学名称,以norank作为标记;6种细菌属于未培养细菌,以uncultured表示。属水平分类细菌包括大量未分类和未培养细菌,给研究细菌的生态功能带来困难。硝化螺菌属(Nitrospira)是丰度最高的属,平均丰度为10.2%,也是硝化螺旋菌门(Nitrospirae)的唯一属水平分类,是泥滩带(S1)、苔草带(S3)和芦苇带(S4)相对丰度最大的属,也是苔草-虉草带(S2)相对丰度第2大的属,但在藜蒿带(S5)相对丰度很小(0.4%)。厌氧绳菌科的一部分属水平未培养细菌(Anaerolineaceae_uncultured)平均丰度为5.3%,占绿弯菌门(Chloroflexi)的33.2%,在泥滩带(S1)的相对丰度最高(13.4%)。酸杆菌科中的一部分属水平未培养细菌(Acidobacteriaceae_(Subgroup_1) _uncultured)平均丰度为5.0%,占酸杆菌门(Acidobacteria)的55.4%,沿湖面至坡地呈增大趋势,是藜蒿带(S5)相对丰度最大的属。乳球菌属(Lactococcus)平均丰度为4.6%,在厚壁菌门(Firmicutes)中占52.5%,是苔草-虉草带(S2)相对丰度最大的属。亚硝化单胞菌科的一部分属水平未培养细菌(Nitrosomonadaceae_uncultured)平均丰度为3.8%,占变形菌门(Proteobacteria)的13.0%,主要分布在苔草-虉草带(S2)、苔草带(S3)和芦苇带(S4)。
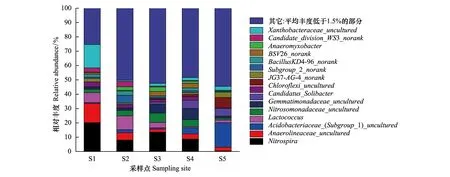
图4 鄱阳湖湿地土壤细菌属水平分类Fig.4 Relative abundances of bacterial genera in Poyang Lake wetland soils
2.3 细菌群落与土壤化学指标的关系
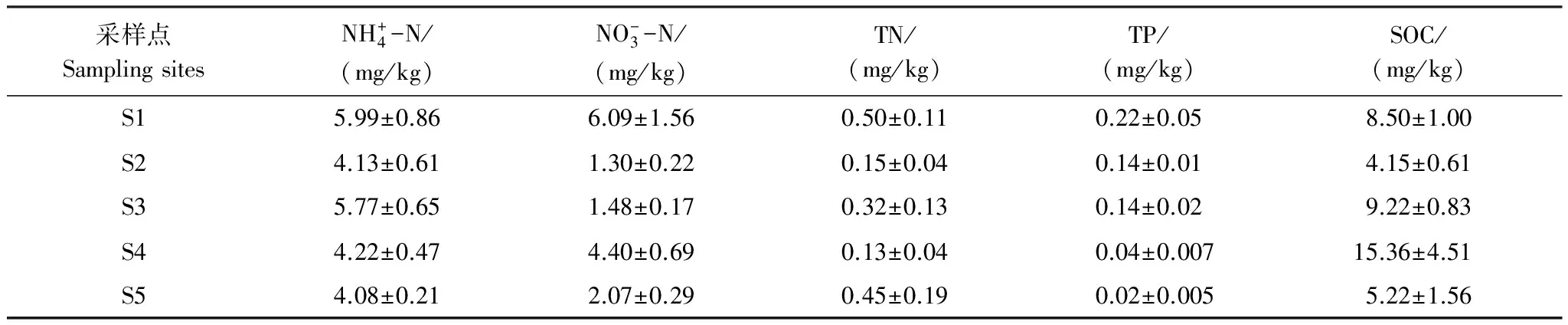
表2 采样点土壤化学指标
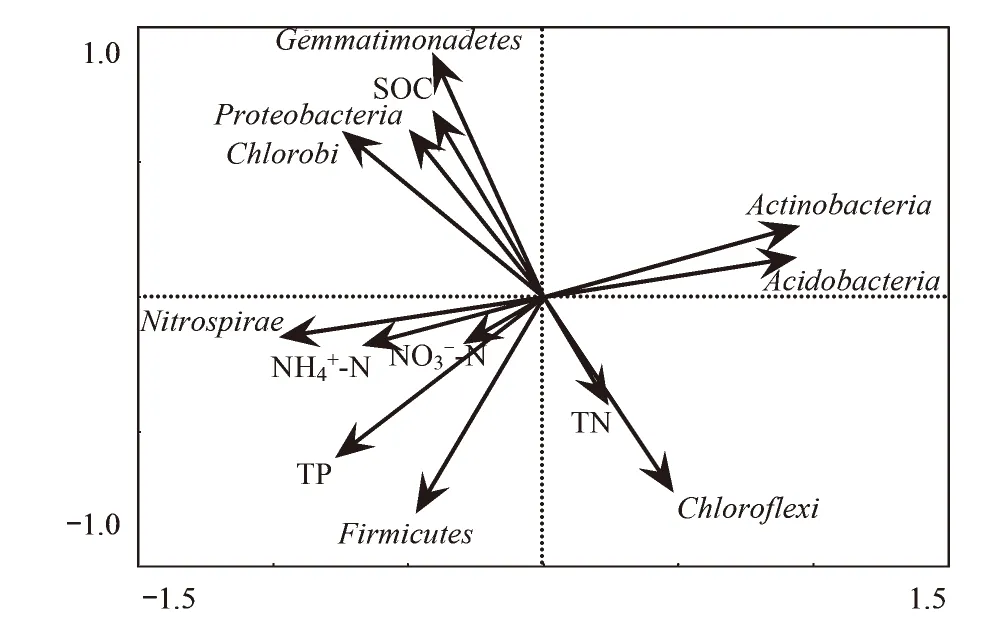
图5 鄱阳湖湿地土壤细菌门分类群落与土壤化学指标的冗余分析 Fig.5 Redundancy analysis of bacterial phyla and chemical parameters in Poyang Lake wetland soils
3 讨论
物种多样性是维持生态系统正常功能的前提条件[19],细菌群落的丰富性和多样性使湿地在营养盐循环、有机物降解、重金属形态转化和温室气体排放等方面起着重要的生态功能[20- 22]。本次研究区的5种湿地植物土壤类型中,土壤细菌群落丰度和多样性的大小具有一致:苔草带>苔草-虉草带>芦苇带>泥滩带>藜蒿带(表 1),即沿湖面至坡地的土壤断面中,中间位置的土壤具有更高的菌群丰度和多样性;同时空间位置相近的土壤细菌群落结构具有更大的相似性(图 2)。由于鄱阳湖水位的季节性变化,5种土壤的淹水时间和淹水频率有较大差别,采样点所处的藜蒿带除在特大洪水年份一般不会被淹水,泥滩带一年大部分时间都处于淹水状态,其它3种土壤则随鄱阳湖水位涨落处于季节性干湿交替状态。很多研究表明水文条件的变化对土壤微生物群落有重要影响[23- 24],Foulquier等[25]研究认为干湿交替的环境有利于一部分细菌的生长,永久淹没区和干湿交替区土壤的微生物群落存在结构性差异。Yu等[26]对沿水文梯度分布的不同湿地研究发现,平均水位和水位变化都会影响土壤微生物群落,但由水文变化引起的沉积作用导致的土壤基质差异对湿地微生物群落的影响更大。鄱阳湖湿地的季节性水位变化及其引起的土壤基质差异可能对土壤细菌群落结构有着重要影响。除水文条件外,不同植被的根系特征,如附着面、根系分泌物和限际氧化还原条件也可能对土壤细菌群落造成影响[27- 28]。
酸杆菌门是藜蒿带相对丰度最高的门,也是研究区土壤平均相对丰度第2高的门;酸杆菌门大多为未培养细菌,目前研究较少,但广泛存在于土壤中,16S rRNA基因克隆文库的分析表明酸杆菌门在典型土壤的丰度约为20%[38],与本文5种土壤的平均丰度(16.7%)接近;Zeglin等[39]分析了河流冲积物沿水分梯度的细菌群落变化,发现酸杆菌门在干旱土壤的丰度较高,这与本次研究结果类似,酸杆菌门相对丰度沿湖面至坡地相对丰度大致呈不断增大的趋势,在很少被淹没的藜蒿带达到最大。绿弯菌门是泥滩带土壤相对丰度最高的门,也是研究区平均相对丰度第3高的门。绿弯菌门与SOC呈负相关性(图 5),绿弯菌门是一类通过光合作用,以CO2为碳源产生能量的细菌[40];绿弯菌门的这种光合特性可能使其在SOC含量较低的土壤中具有竞争优势。另外有研究表明绿弯菌门在水位频繁变化的潮间带土壤含量较高[36,41],泥滩带较高的绿弯菌门丰度可能也与其频繁的水位变化有关。硝化螺旋菌是研究区土壤平均相对丰度第4高的门,只包含了硝化螺菌属一个属,硝化螺菌属也是湿地土壤平均丰度最高的属分类单元;一般湿地土壤的硝化螺旋菌门丰度很小(<1%)[36],远低于本次研究结果(13.5%),同时亚硝化单胞菌科的一部分属水平未培养细菌也是变形菌门丰度最大的属分类群落(图 4);这些都表明氮素循环在鄱阳湖湿地生态功能中可能占有重要地位。
4 结论
(1)高通量测序结果表明鄱阳湖湿地5个土壤样品中的2 072个OTUs分属于37个门,86个纲,176个目,294个科,401个属,718个种,样品中包括大量未分类和未培养细菌。植被土壤细菌群落丰度与多样性的排序是一致的:苔草带>苔草-虉草带>芦苇带>泥滩带>藜蒿带。
(2)变形菌门(Proteobacteria,30.0%)是鄱阳湖湿地平均相对丰度最高的门,其次为酸杆菌门(Acidobacteria,16.7%),绿弯菌门(Chloroflexi,16.5%),硝化螺旋菌门(Nitrospirae,10.2%),厚壁菌门(Firmicutes,7.5%)。硝化螺菌属(Nitrospira)是第一大属分类水平细菌群落。

[1] Faulwetter J L, Gagnon V, Sundberg C, Chazarenc F, Burr M D, Brisson J, Camper A K, Stein O R. Microbial processes influencing performance of treatment wetlands: a review. Ecological Engineering, 2009, 35(6): 987- 1004.
[2] 刘驰, 李家宝, 芮俊鹏, 安家兴, 李香真. 16S rRNA基因在微生物生态学中的应用. 生态学报, 2015, 35(9): 2769- 2788.
[3] Zhang J X, Yang Y Y, Zhao L, Li Y Z, Xie S G, Liu Y. Distribution of sediment bacterial and archaeal communities in plateau freshwater lakes. Applied Microbiology and Biotechnology, 2015, 99(7): 3291- 3302.
[4] 胡安谊, 李姜维, 杨晓永, 王弘杰, 于昌平. 宁波三江口水域原核生物群落结构分析. 环境科学, 2015, 36(7): 2487- 2495.
[5] 唐婧, 徐小蓉, 商传禹, 牛晓娟, 张习敏, 乙引. 南明河城区河段细菌多样性与环境因子的关系. 微生物学报, 2015, 55(8): 1050- 1059.
[6] Wang Z, Yang Y Y, He T, Xie S G. Change of microbial community structure and functional gene abundance in nonylphenol-degrading sediment. Applied Microbiology and Biotechnology, 2015, 99(7): 3259- 3268.
[7] 寇文伯, 黄正云, 张杰, 刘倩纯, 刘芳鹏, 刘以珍, 吴兰. 鄱阳湖湖泊细菌群落组成及结构——以松门山为例. 生态学报, 2015, 35(23): 7608- 7614.
[8] 白晓旭, 史荣久, 尤业明, 盛华芳, 韩斯琴, 张颖. 河南宝天曼不同林龄与林型森林土壤的细菌群落结构与多样性. 应用生态学报, 2015, 26(8): 2273- 2281.
[9] Ligi T, Oopkaup K, Truu M, Preem J K, Nõlvak H, Mitsch W J, Mander Ü, Truu J. Characterization of bacterial communities in soil and sediment of a created riverine wetland complex using high-throughput 16S rRNA amplicon sequencing. Ecological Engineering, 2014, 72: 56- 66.
[10] 李晨华, 张彩霞, 唐立松, 熊正琴, 王保战, 贾仲君, 李彦. 长期施肥土壤微生物群落的剖面变化及其与土壤性质的关系. 微生物学报, 2014, 54(3): 319- 329.
[11] Shao M Q, Jiang J H, Guo H, Zeng B B. Abundance, distribution and diversity variations of wintering water birds in Poyang lake, Jiangxi province, china. Pakistan Journal of Zoology, 2014, 46(2): 451- 462.
[12] 胡振鹏, 葛刚, 刘成林, 陈伏生, 李述. 鄱阳湖湿地植物生态系统结构及湖水位对其影响研究. 长江流域资源与环境, 2010, 19(6): 597- 605.
[13] Hu Y X, Huang J L, Du Y, Han P P, Wang J L, Huang W. Monitoring wetland vegetation pattern response to water-level change resulting from the Three Gorges Project in the two largest freshwater lakes of China. Ecological Engineering, 2015, 74: 274- 285.
[14] 王晓龙, 徐立刚, 姚鑫, 白丽, 张奇. 鄱阳湖典型湿地植物群落土壤微生物量特征. 生态学报, 2010, 30(18): 5033- 5042.
[15] 张杰, 胡维, 刘以珍, 葛刚, 吴兰. 鄱阳湖湿地不同土地利用方式下土壤微生物群落功能多样性. 生态学报, 2015, 35(4): 965- 971.
[16] Liu L X, Xu M, Qiu S, Shen R C. Spatial patterns of benthic bacterial communities in a large lake. International Review of Hydrobiology, 2015, 100(3/4): 97- 105.
[17] Yu C, Zhang J, Wu L, Liu Y Z, Ge G. Effects of heavy metal and nutrients on benthic microbial communities in freshwater sediment of Poyang Lake (China). Journal of Residuals Science & Technology, 2015, 12(2): 105- 111.
[18] Dennis K L, Wang Y, Blatner N R, Wang S, Saadalla A, Trudeau E, Roers A, Weaver C T, Lee J J, Gilbert J A, Chang E B, Khazaie K. Adenomatous polyps are driven by microbe-instigated focal inflammation and are controlled by IL- 10-producing T cells. Cancer Research, 2013, 73(19): 5905- 5913.
[19] Ansola G, Arroyo P, de Miera L E. Characterisation of the soil bacterial community structure and composition of natural and constructed wetlands. Science of the Total Environment, 2014, 473- 474: 63- 71.
[20] Kosolapov D B, Kuschk P, Vainshtein M B, Vatsourina A V, Wießner A, Kästner M, Müller R A. Microbial processes of heavy metal removal from carbon‐deficient effluents in constructed wetlands. Engineering in Life Sciences, 2004, 4(5): 403- 411.
[21] Martins G, Terada A, Ribeiro D C, Corral A M, Brito A G, Smets B F, Nogueira R. Structure and activity of lacustrine sediment bacteria involved in nutrient and iron cycles. FEMS Microbiology Ecology, 2011, 77(3): 666- 679.
[22] Conrad R. Soil microorganisms as controllers of atmospheric trace gases (H2, CO, CH4, OCS, N2O, and NO). Microbiological Reviews, 1996, 60(4): 609- 640.
[23] Rees G N, Watson G O, Baldwin D S, Mitchell A M. Variability in sediment microbial communities in a semipermanent stream: impact of drought. Journal of the North American Benthological Society, 2006, 25(2): 370- 378.
[24] Mentzer J L, Goodman R M, Balser T C. Microbial response over time to hydrologic and fertilization treatments in a simulated wet prairie. Plant and Soil, 2006, 284(1/2): 85- 100.
[25] Foulquier A, Volat B, Neyra M, Bornette G, Montuelle B. Long-term impact of hydrological regime on structure and functions of microbial communities in riverine wetland sediments. FEMS Microbiology Ecology, 2013, 85(2): 211- 226.
[26] Yu S, Ehrenfeld J G. Relationships among plants, soils and microbial communities along a hydrological gradient in the New Jersey Pinelands, USA. Annals of Botany, 2010, 105(1): 185- 196.
[27] Zhang J, Wang Q, Fan J L, Xie H J, Liu C, Liang S, Hu Z, Yang Z C, Zhao C C. Comparisons of microbial abundance and community among different plant species in constructed wetlands in summer. Ecological Engineering, 2015, 82: 376- 380.
[28] Faulwetter J L, Burr M D, Parker A E, Stein O R, Camper A K. Influence of season and plant species on the abundance and diversity of sulfate reducing bacteria and ammonia oxidizing bacteria in constructed wetland microcosms. Microbial Ecology, 2013, 65(1): 111- 127.
[29] 李靖宇, 杜瑞芳, 赵吉. 乌梁素海富营养化湖泊湖滨湿地过渡带细菌群落结构的高通量分析. 微生物学报, 2015, 55(5): 598- 606.
[30] Yun J L, Ju Y W, Deng Y C, Zhang H X. Bacterial community structure in two permafrost wetlands on the Tibetan Plateau and Sanjiang plain, China. Microbial Ecology, 2014, 68(2): 360- 369.
[31] Deng Y C, Cui X Y, Hernández M, Dumont M G. Microbial diversity in hummock and hollow soils of three wetlands on the Qinghai-Tibetan plateau revealed by 16S rRNA pyrosequencing. PLoS One, 2014, 9(7): e103115.
[32] Yu Y, Wang H, Liu J, Wang Q, Shen T L, Guo W H, Wang R Q. Shifts in microbial community function and structure along the successional gradient of coastal wetlands in Yellow River Estuary. European Journal of Soil Biology, 2012, 49: 12- 21.
[33] Jiang X T, Peng X, Deng G H, Sheng H F, Wang Y, Zhou H W, Tam N F Y. Illumina sequencing of 16S rRNA tag revealed spatial variations of bacterial communities in a mangrove wetland. Microbial Ecology, 2013, 66(1): 96- 104.
[34] Dedysh S N, Ricke P, Liesack W. NifH and NifD phylogenies: an evolutionary basis for understanding nitrogen fixation capabilities of methanotrophic bacteria. Microbiology, 2004, 150(Pt 5): 1301- 1313.
[35] Madigan M T, Martinko J M, Parker J. Brock Biology of Microorganisms. Upper Saddle River, NJ: Prentice Hall, 1997.
[36] Lv X F, Yu J B, Fu Y Q, Ma B, Qu F Z, Ning K, Wu H F. A Meta-Analysis of the bacterial and archaeal diversity observed in wetland soils. The Scientific World Journal, 2014, 2014: Article ID 437684.
[37] Song H, Li Z, Du B, Wang G, Ding Y. Bacterial communities in sediments of the shallow Lake Dongping in China. Journal of Applied Microbiology, 2012, 112(1): 79- 89.
[38] Janssen P H. Identifying the dominant soil bacterial taxa in libraries of 16S rRNA and 16S rRNA genes. Applied and Environmental Microbiology, 2006, 72(3): 1719- 1728.
[39] Zeglin L H, Dahm C N, Barrett J E, Gooseff M N, Fitpatrick S K, Takacs-Vesbach C D. Bacterial community structure along moisture gradients in the parafluvial sediments of two ephemeral desert streams. Microbial Ecology, 2011, 61(3): 543- 556.
[40] Klatt C G, Liu Z F, Ludwig M, Kühl M, Jensen S I, Bryant D A, Ward D M. Temporal metatranscriptomic patterning in phototrophic Chloroflexi inhabiting a microbial mat in a geothermal spring. The ISME Journal, 2013, 7(9): 1775- 1789.
[41] Wang Y, Sheng H F, He Y, Wu J Y, Jiang Y X, Tam N F Y, Zhou H W. Comparison of the levels of bacterial diversity in freshwater, intertidal wetland, and marine sediments by using millions of Illumina tags. Applied and Environmental Microbiology, 2012, 78(23): 8264- 8271.
High throughput sequencing analysis of bacterial communities in soils of a typical Poyang Lake wetland
WANG Peng1,2,*, CHEN Bo1,2,ZHANG Hua1,2
1KeyLaboratoryofPoyangLakeWetlandandWatershedResearch,MinistryofEducation,JiangxiNormalUniversity,Nanchang330022,China2SchoolofGeographyandEnvironment,JiangxiNormalUniversity,Nanchang330022,China

Poyang Lake wetland; high throughput sequencing; bacterial diversity; bacterial community structure
江西省自然科学基金(20151BAB213035);鄱阳湖湿地与流域研究教育部重点实验室(江西师范大学)主任开放基金(ZK2013009);江西省教育厅科技计划项目(GJJ14267);江西省重大生态安全问题监控协同创新中心资助项目(JXS-EW-00)
2015- 10- 05;
日期:2016- 07- 13
10.5846/stxb201510052000
*通讯作者Corresponding author.E-mail: wangpengjlu@outlook.com
王鹏,陈波,张华.基于高通量测序的鄱阳湖典型湿地土壤细菌群落特征分析.生态学报,2017,37(5):1650- 1658.
Wang P, Chen B,Zhang H.High throughput sequencing analysis of bacterial communities in soils of a typical Poyang Lake wetland.Acta Ecologica Sinica,2017,37(5):1650- 1658.
