程序升温表面反应技术在固体催化剂上的应用(上)
2017-05-12黄文氢
满 毅,黄文氢,陈 松
(中国石化 北京化工研究院,北京 100013)
程序升温表面反应技术在固体催化剂上的应用(上)
满 毅,黄文氢,陈 松
(中国石化 北京化工研究院,北京 100013)
简要介绍了程序升温表面反应(TPSR)技术的基本原理、TPSR的装置及实验条件的选择,重点综述了近十年来TPSR技术在烯烃和烷烃的重整氧化、费托合成、NOx的催化还原、醇醛的氧化还原、CO的氧化、硫氮氯化合物的分解等催化研究中的应用情况,并探讨了TPSR技术的发展趋势。
程序升温表面反应;固体催化剂;石油化工;环境污染物治理
程序升温技术是一项研究固体催化剂的重要技术,广泛应用于固体催化剂的测试表征[1-2],具有简易可行、实时监测等特点,主要原理是:固体催化剂或预吸附某些气体的固体催化剂,在载气中以一定的升温速率加热,检测流出气体的组成和浓度的变化或固体(表面)物理性质和化学性质的变化。按照分析目的的不同,程序升温技术主要分为程序升温脱附(TPD)、程序升温还原(TPR)、程序升温氧化(TPO)、程序升温表面反应(TPSR)。其中,TPSR研究的是催化剂表面吸附的物种在程序升温过程中的脱附及其与其他物种发生反应或自身发生变化的过程,与对某一种或几种吸附物种仅进行脱附分析的TPD实验不同,TPSR可在反应条件下得到催化剂表面上吸附物种与其他物种发生反应的重要信息。1984年,王晓鸣[3]对TPSR及其在催化研究中的应用进行了介绍,包括基本原理和实验方法,并对TPSR的某些动力学参数进行数学推导,对采用TPSR研究表面吸附态和反应机理等进行了举例说明。由于TPSR过程中涉及的物种较多,单一的检测器(如TCD)很难进行检测。近年来,随着质谱、红外与多检测器化学吸附仪联用技术的进一步发展,联用技术更多地应用在催化剂TPSR的表征和分析中,并可进一步研究催化反应的活性中心及反应动力学等。
本文简要介绍了TPSR技术的基本原理、装置及实验条件的选择,综述了近年来TPSR技术在烯烃和烷烃的重整氧化、费托合成、NOx催化还原、醇醛氧化还原、CO氧化、硫氮氯化合物分解等催化过程中的应用,并在此基础上提出了TPSR技术今后的发展趋势。
1 仪器结构
TPSR实验通常需要在动态(流动气体)条件下进行,包括以下几个部分:气路系统、温控系统、样品池、检测系统[4]。另外,根据实验需要还可选配液体进料器、气液分离器、超高真空系统等配件。
1.1 气路系统
仪器气路通常配备准备气路、反应气路、脉冲气路等,管路通常为耐腐蚀的不锈钢材质,管路长度尽可能短以减小死体积。气路中通常配备六通阀、开关阀,以便实现多种气体或气液混合。
1.2 温控系统
温控系统包括对样品池进行恒温、升温和降温,对样品池的温度进行控制;同时也包括对管路和检测器进行加热,以保证恒温条件下某些低沸点的气体不会冷凝和吸附在管路中。
1.3 样品池
样品池用于放置或固定试样,形状没有限制,一般为U型管,有时也会用到薄片或立方体形状的样品池。对于常压表面反应,样品池的材质一般为石英;对于高压表面反应,样品池的材质一般为不锈钢。
1.4 检测系统
检测系统用于记录TPSR的信号,包括TCD、质谱检测器、化学发光检测器、红外检测器、FID等。根据检测物质和检测目的的不同,可单独使用以上任意一种检测器或联用多种检测器。
TCD的检测原理是检测热导值。TCD的灵敏度高,定量效果好,可以检测几乎所有组分,对检测物不会造成破坏。但应用于TPSR时,无法分辨多种物质。
质谱检测器的检测原理是检测分子或碎片峰质量数。质谱检测器的灵敏度极高,定量效果中等,可以检测几乎所有组分,会对检测物造成破坏。质谱检测器可以检测不同组分(质量数)随温度的变化,但应用于TPSR时,一般信号响应时间较长,普通四极杆质谱无法区分相同质量数。
化学发光检测器的检测原理是检测物质发射出的光子。化学发光检测器的灵敏度极高,定量效果极好,可以检测硫、氮等化合物,但会对检测物造成破坏。化学发光检测器的选择性好,对硫、氮等为摩尔响应,但应用于TPSR时,只对指定物质有响应,无法区分含相同元素的不同物质。
红外检测器的检测原理是检测化学键或官能团的红外吸收。红外检测器的灵敏度中等,可以检测有红外吸收的物质,不会对检测物造成破坏。红外检测器的优点是可以区分不同官能团,但应用于TPSR时,定量效果差,无法区分具有相同官能团的不同物质。
FID的检测原理是检测有机物燃烧生成的离子流。FID的灵敏度极高,定量效果极好,可以检测碳氢化合物,会对检测物造成破坏。FID的优点是适用有机物的检测,灵敏度高,线性范围广,价格便宜。但应用于TPSR时,FID只对可燃烧碳氢化合物有响应,属于破坏性检测器,无法区分不同物质。
1.5 液体进料器
液体进料器用于常温下将液态的物质引入到样品池中,常用的有蒸气发生器和高压液相泵。蒸气发生器是通过惰性气体(He、Ar)或反应气体将一定温度下的液体饱和蒸气带入样品池中;高压液相泵是通过外压将液体以一定流速注入样品池中。
1.6 气液分离器
气液分离器用于对气、液产物进行实时分离和采集。
1.7 超高真空系统
可以利用超高真空系统对模型催化剂进行测试,在模型催化剂特定的晶面上进行吸附和表面反应,观察不同晶面活性位的不同。
2 TPSR的实验条件
设计合适的实验条件是得到合理分析结果的重要前提。实验条件一般包括预处理、吸附、吹扫/抽真空、表面反应这四个步骤,其中,前三个步骤可根据具体实验要求省略。
2.1 预处理
预处理是对试样表面进行净化和活化,可在惰性气氛(He、Ar)或氧化、还原气氛(H2、O2)中进行处理,处理温度以不破坏试样并充分活化和净化试样为宜,必要时可先对试样进行热重、TPR等实验来确定合适的处理温度。
2.2 吸附
有脉冲吸附和连续吸附两种方式,通过脉冲次数或连续吸附时间来控制吸附量。
2.3 吹扫/抽真空
采用惰性气体吹扫或抽真空的方式,将试样上物理吸附和管路残留的吸附质去除。尽可能采用长时间的吹扫或抽真空,以保证完全脱除。
2.4 表面反应
在程序升温的过程中可通入惰性气体、一种或多种反应气体,为保证信号的采集效果,最好在基线稳定后再开始升温。有时根据实验需要,可以利用吸附质的同位素进行吸附或表面反应实验。
对中国石化北京化工研究院制备的Pd/Al2O3催化剂的乙炔加氢性能进行研究,按Pd含量递增的顺序将催化剂分别编号为MS03P,MS13P3,MS22P4。将催化剂装入试样管中,在Ar吹扫下以10 ℃/min的速率升温至350 ℃,用H2还原1 h,再用Ar吹扫1 h,降至室温,连续通入C2H2-H2混合气,基线稳定后,以10 ℃/min的速率升温至350 ℃,用质谱检测H2(m/z= 2)、C2H2(m/z= 26)和C2H2(m/z= 27),结果如图1所示。

图1 不同Pd/Al2O3催化剂的C2H2-H2-TPSR实验结果Fig.1 C2H2-H2-TPSR result on different Pd/Al2O3catalysts.
从图1可看出,在室温下C2H2和H2在3种Pd/Al2O3催化剂上可少量反应生成C2H4,随温度的升高,从70 ℃开始,C2H2转化率逐渐变大,80 ℃后C2H2转化率保持稳定。对于3种不同的Pd催化剂,MS13P3和MS22P4的C2H2初始转化温度低于MS03P,表明C2H2和H2更易在MS13P3和MS22P4上发生反应。
3 TPSR技术在催化领域的应用
3.1 烯烃烷烃重整氧化
烯烃和烷烃是石油化工重要的原料和产物,对其进行重整氧化可提高汽油质量,并产生有用的副产物H2,将简单的烃转化为更实用的醛和醚等。通过TPSR技术可以研究反应的中间体、氧物种、积碳及动力学性质等。
Lwin等[5]通过C3D6/C3H6-TPSR研究了ReOx/ Al2O3催化剂的烯烃反应机理和动力学性质,通过H的同位素D进行表面反应研究,采用质谱进行了检测,结果如图2所示。由图2可见,催化剂上不同的C3物种的出峰温度不同,且不同的C3物种在不同温度下的比例不同。同时,丙烯的复分解反应在不同温度下的反应途径不同:在低于100 ℃时,生成Re*==CH2和Re*==CHCH3中间体,且与气相丙烯存在吸脱附平衡;在高于100 ℃时,表面生成C3==π混合物,且表面重组的Re*==CH2和Re*==CHCH3也有助于丙烯在高温下发生复分解反应。
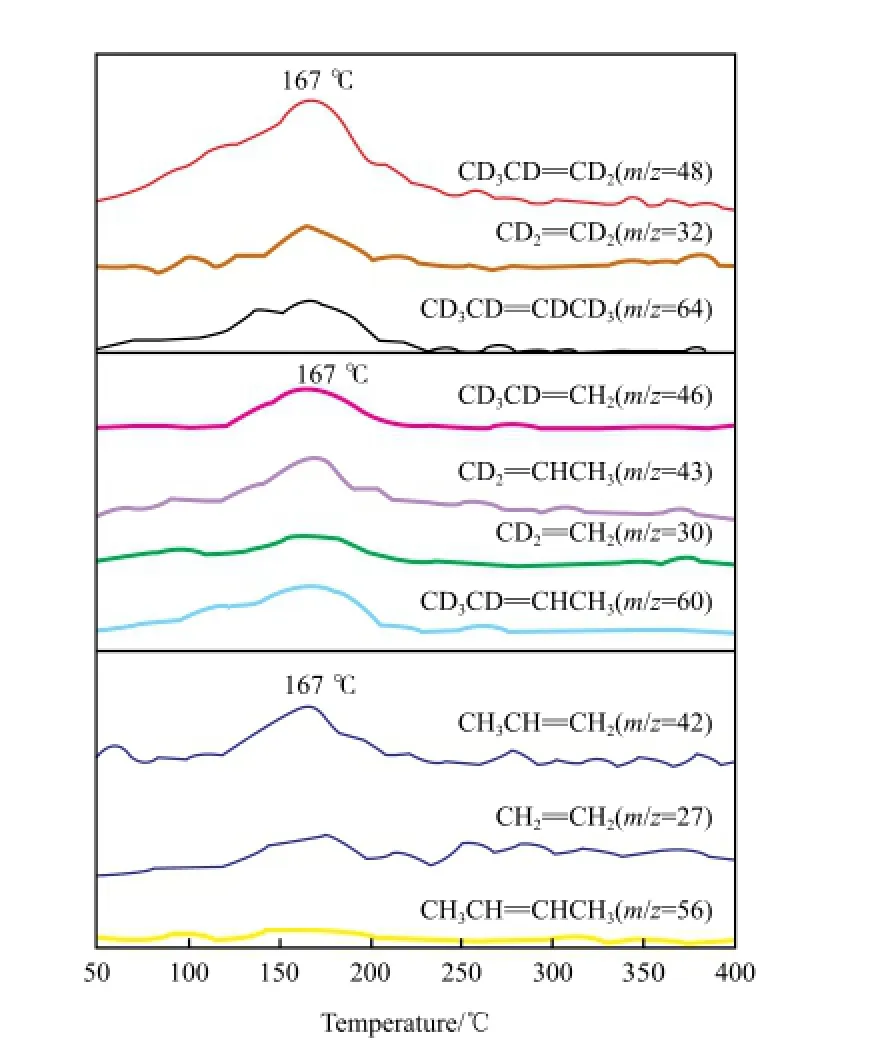
图2 Ar/C3D6(30 ℃)/C3H6(100 ℃)-TPSR得到的同位素标记的反应产物的质谱信号变化[5]Fig.2 MS signals of isotope-labeled reaction products from Ar/C3D6(30 ℃)/C3H6(100 ℃)-TPSR[5].
Guimarães等[6]通过C3H8-O2-TPSR研究了Pd/ CeO2/Al2O3催化剂上丙烷在有O2条件和有H2O条件下的部分氧化。结果表明,催化剂中加入CeO2影响了氧化过程,在氧气充足的条件下,产物只有CO2;当氧气被消耗后,除了生成CO2,也会生成CO和H2;在无氧条件下,加入CeO2会生成更多的H2。Zhao等[7]采用C3H6-TPSR实验通过18O2同位素测定了催化剂在丙烯氧化制丙烯醛的过程中气相分子氧和晶格氧的作用。结果表明,在预吸附丙烯的条件下,丙烯醛的生成仅仅发生在气相分子氧存在的时候;在通入18O2同位素时,含18O2同位素的丙烯醛并没有生成,说明是V2O5/Nb2O5中的氧参与了反应。对预吸附的丙烯分别用He和O2进行程序升温[8],在无O2条件下,化学吸附的丙烯在升温至500 ℃的范围内没有丙烯醛生成;而在有O2条件下,化学吸附的丙烯分别在206 ℃和349 ℃生成丙烯醛,实验结果遵循了Langmuir-Hinshelwood反应机理,即两种反应物必须同时存在并吸附在催化剂表面才会发生反应。通过峰温,根据Redhead方程[9]计算得到反应的活化能为136.9 kJ/mol。
Wang等[10]用H2-TPSR、CO2-TPSR和程序升温氧化反应研究了甲烷重整过程中的碳中间体,检测到了在Ni-CaO-ZrO2催化剂表面有3种碳物种,分别是Cα,Cβ,Cγ,在反应中,3种表面碳物种与催化剂表面的相互作用是不同的,导致了催化剂的性能不同。Guo等[11]利用CO2-TPSR研究了甲烷重整过程中的积碳,对比了浸渍法合成的Ni/Al2O3催化剂与用等离子体辐照Mn改性的Ni/Al2O3催化剂,后者有利于积碳的消除,具有更好的抗积碳性能。Zhu等[12]通过CH4-TPSR研究了不同金属改性的Mo2C/Al2O3催化剂催化甲烷制合成气的效果。结果表明,镍的加入促进了甲烷部分氧化制合成气;在反应初期,铜表现出较弱的催化活性,不利于催化;钾的加入也不利于催化。
Xiao等[13]通过CH4-TPSR研究了AFeO3(A= La,Nd,Eu)催化剂上甲烷部分氧化制合成气过程中的氧物种,反应中AFeO3催化剂提供了氧物种,这些氧物种起到唯一的氧化作用,而气相中的氧并没有直接参与反应,消耗的氧物种在氧气气氛下重新生成。3种催化剂中LaFeO3具有最多的活性氧物种,同时也有相对最好的催化活性。Pantaleo等[14]通过CH4-TPSR研究了Ni负载的CeO2催化剂上的晶格氧,催化剂表面的积碳可与CeO2中的晶格氧作用生成CO,从而去除积碳;而NiO的加入使其与CeO2相互作用产生更多的晶格氧,从而更有利于消除积碳,有利于甲烷部分氧化。Álvarez-Galván等[15]采用C3H8-TPSR研究了LaCoO3和Ru/LaCoO3催化剂,催化剂分别经750 ℃下10%(φ)H2处理2 h、400 ℃下10%(φ)O2处理2 h、600 ℃下10%(φ)O2处理2 h后的反应历程。结果表明,LaCoO3和Ru/LaCoO3催化剂上都会生成CO、CO2、H2、CH4和积碳,反应机理均需晶格氧和在Co0(金属态)上生成的H2的参与。无论经过氧化还是还原处理,Ru/LaCoO3都具有更好的活性,且积碳量(2.13 mmol/g)小于LaCoO3(3.65 mmol/g)。宋国华等[16]同样采用C3H8-TPSR研究了丙烷氧化脱氢反应中不同氧化硅载体负载的钒催化剂的结构和催化性能。结果表明,负载型钒氧化物催化剂存在两种晶格氧,活性较高的晶格氧在低温使丙烷完全氧化,且高活性的晶格氧与催化剂存在键合强度的差异;活性较低的晶格氧在高温使丙烷氧化脱氢为丙烯。
3.2 醇醛氧化还原
醇和醛是化工生产中重要的原料和中间产物,对其进行氧化还原等可得到附加值更高的产品或中间体。由于大部分醇和醛在常温下都是液体,所以需要用到液体进料器或配置低浓度的标气。
Omar等[17]采用O2-TPSR在不同的吸附和表面反应条件下研究了2-丁醇在Pd/AlPO4催化剂上的氧化反应,同时用FID和TCD检测产物。实验结果表明,不同的吸附和表面反应条件可以导致不同的产物,催化剂表面上的氧有利于醇盐物种的形成;催化剂表面活性位形成PdO/Pd,PdO的存在有利于生成甲基乙基酮。
Li等[18]通过乙醇/O2-TPSR研究了乙醇在CeO2纳米颗粒催化剂上的选择性氧化过程。实验结果表明,乙酸根和乙氧基是在CeO2上产生的表面物种;由于CeO2的3种不同形态(八面体、立方体、纳米棒)导致它有不同的表面酸性、不同的氧空位数量和吸附位的几何形状,产物乙醛、乙烯、CO2的比例也不同。Gambaro等[19]用异丙醇-TPSR来确定杂多酸H6P2W18O62·nH2O的表面酸量,异丙醇可吸附在表面和体相中,由于材料的伪液相性质,具有高Brönsted和Lewis酸性的杂多酸催化剂与单层负载的WOx/TiO2催化剂相比,可在较低温度下(~100 ℃)催化反应。另外,含水的杂多酸比脱水酸有更多的活性位,因此含水的杂多酸产生的丙烯是脱水酸的2倍。
Mann等[20]采用TPSR研究了乙醛在3种不同结构CeO2纳米晶上(立方体、八面体、纳米线)的吸附和表面反应,用四极杆质谱进行检测,得到了不同结构CeO2纳米晶上产物随温度的变化(如图3所示)。由图3可见,CeO2表面不同的结构决定了乙醛在脱附过程中是否发生变化或发生还原、耦合、C—C键的断裂。乙醛消耗量的大小顺序为:纳米线>立方体>八面体,乙醇选择性的大小顺序为:纳米线≈立方体>>八面体。选择性的差异可归结于催化剂表面的碱性、缺陷、表面原子的配位数和还原能力。

图3 CeO2立方体(A,B)、纳米线(C,D)和八面体(E,F)上乙醛表面反应产物的质谱信号变化[20]Fig.3 TPSR profiles under a constant acetaldehyde stream on CeO2cubes(A,B),wires(C,D),and octahedra(E,F)[20].
Chen等[21]研究了不同Ag负载量下Ag/MCM-41催化剂上甲醛的吸附和表面反应,分别在Ar和O2氛围下进行程序升温。结果表明,Ag含量的不同会显著影响甲醛的吸附和表面反应;在Ag含量小于8%(w)时,甲醛的脱附温度会随着Ag含量的增加而降低,甲醛对O2的表面反应活性增大;当Ag含量超过8%(w)时,由于Ag的聚集,甲醛的脱附温度会随着Ag含量的增加而升高,甲醛对O2的表面反应活性变低。研究结果表明,合适的Ag负载量和Ag的分散在低温下会提高反应活性。
Molinari等[22]采用CH3OH-TPSR研究了系列磷钼氧钒催化剂的甲醇氧化反应,采用质谱检测了不同组分随温度的变化。对不同的催化剂,H2CO的出峰位置不同,随着催化剂中钒含量的增加,出峰位置逐渐降低,钒的加入引起了催化剂结构的变化,从而导致反应温度变化。Nakka等[23]同样采用CH3OH-TPSR研究了钒钨酸盐的表面和体相性质,氧化钒在Keggin结构内和结构外均有生成,但外表面的氧化钒的活性明显小于结构内氧化钒的活性。Wachs等[24]采用CH3OH-TPSR研究了混合金属氧化物Mo-V-Te-Nb-O表面活性位,确定了不同催化剂的峰温:V(173~188 ℃),Mo(189~196 ℃),Te(260~430 ℃),Nb(300 ℃),再通过峰温计算出反应活化能,深入讨论了峰温与M—O—S价键之间的关系。Guerrero-Pérez等[25]研究了Sb-V-O氧化物活性表面的数量和化学性质(氧化还原性和酸性),锑酸盐含有很少量的活性氧化态和酸性,氧化铝负载的钒氧化物有很多的表面氧化位和少量酸性,但锑酸盐负载的钒氧化物却具有更多的氧化位,进一步研究发现,其中含有VSbO4相,使得表面具有更多的氧化钒。
Routray等[26]根据CH3OH-TPSR实验计算了金属钒酸盐催化剂MVO4(M=Al,Fe,Nb,Cr,Zn,Ni,Cu)催化下反应的活化能和速率常数。结果表明,计算得到的活化能和速率常数与M==O键的长度和强度无关,说明CH3OH的氧化需要在MOx催化剂表面断裂C—H键,与M==O键的断裂无关。Lee等[27]通过CH3OH-TPSR用Redhead公式计算了SiO2负载的金属氧化物(Al2O3,TiO2,ZrO2,V2O5,CrO3,MoO3,WO3,Re2O7)的活化能。结果表明,第1列过渡金属比第2列和第3列过渡金属更易还原,且氧化还原活性随氧化态的增加而增加。催化剂的活性主要由氧化物自身性质决定。
Chen等[28]研究了Ag在不同载体(MCM-41,SBA-15,NaY,SiO2,TiO2)上的吸附脱附性能和表面反应。结果表明,这几种载体都没有甲醛氧化活性,Ag颗粒具有甲醛氧化活性以及更多的甲醛吸附量。MCM-41和SiO2负载的Ag催化剂有新的甲醛吸附态,在低温下可以活化,有最好的甲醛氧化活性;SBA-15和NaY负载的Ag催化剂的甲醛脱附温度较高,反应性能相对较差;而Ag/TiO2则没有任何活性。吸附在Ag表面的甲醛会形成二氧亚甲基和甲酸盐中间表面态,然后与O2反应生成CO2。Qu等[29]通过研究甲醛在Ag/SBA-15催化剂上的表面反应,比较了嫁接法和传统浸渍法制备的Ag/SBA-15催化剂的差别。在低温下,嫁接法制备的催化剂具有更高的甲醛吸附性能和更高的活性。
[1] 杨锡尧. 固体催化剂的研究方法 第十三章程序升温分析技术(上)[J].石油化工,2001,30(12):952-959.
[2] 杨锡尧. 固体催化剂的研究方法 第十三章程序升温分析技术(下)[J].石油化工,2002,31(1):63-73.
[3] 王晓鸣. 程序升温表面反应及其在催化研究中的应用[J].石油化工,1984,13(2):132-140.
[4] Paul A W,Clyde O. Analytical methods in fi ne partical technology[M].Norcross:Micromeritics Instrument Corporation,1997:239-240.
[5] Lwin S,Wachs I E. Reaction mechanism and kinetics of olefin metathesis by supported ReOx/Al2O3catalysts[J].ACS Catal,2016,6(1):272-278.
[6] Guimarães A L,Dieguez L C,Schmal M. Surface sites of Pd/ CeO2/Al2O3catalysts in the partial oxidation of propane[J].J Phys Chem B,2003,107(18):4311-4319.
[7] Zhao Chunli,Wachs I E. An Operando Raman,IR,and TPSR spectroscopic investigation of the selective oxidation of propylene to acrolein over a model supported vanadium oxide monolayer catalyst[J].J Phys Chem C,2008,112(30):11363-11372.
[8] Zhao Chunli,Wachs I E. Selective oxidation of propylene to acrolein over supported V2O5/Nb2O5catalysts:An in situ Raman,IR,TPSR and kinetic study[J].Catal Today,2006,118(3):332-343.
[9] Redhead P A. Thermal desorption of gases[J].Vacuum,1962,12(4):213-211.
[10] Wang Changzhen,Sun Nannan,Wei Wei,et al. Carbon intermediates during CO2reforming of methane over Ni-CaOZrO2catalysts:A temperature-programmed surface reaction study[J].Int J Hydrogen Energ,2016,41(42):19014-19024.
[11] Guo Fang,Xu Junqiang,Chu Wei. CO2reforming of methane over Mn promoted Ni/Al2O3catalysttreated by N2glow discharge plasma[J].Catal Today,2015,256(1):124-129.
[12] Zhu Quanli,Zhang Bin,Zhao Jun,et al. The effect of secondary metal on Mo2C/Al2O3catalyst for the partial oxidation of methane to syngas[J].J Mol Catal A:Chem,2004,213(2):199-205.
[13] Xiao Pingdai,Ran Jiali,Chang Chunyu,et al. Unsteadystate direct partial oxidation of methane to synthesis gas in a fi xed-bed reactor using AFeO3(A = La,Nd,Eu) perovskitetype oxides as oxygen storage[J].J Phys Chem B,2006,110(45):22525-22531.
[14] Pantaleo G,LaParola V,Deganello F,et al. Ni/CeO2catalysts for methane partial oxidation:Synthesis driven structural and catalytic effects[J].Appl Catal,B,2016,189(15):233-241.
[15] Álvarez-Galván M C,Constantinou D A,Navarro R M,et al. Surface reactivity of LaCoO3and Ru/LaCoO3towards CO,CO2and C3H8:Effect of H2and O2pretreatments[J].Appl Catal,B,2011,102(1/2):291-301.
[16] 宋国华,缪建文,范以宁. 丙烷氧化脱氢不同硅基载体负载钒氧化物催化剂的TPSR表征[J].无机化学学报,2011,27(9):1758-1764.
[17] Omar O,Mohamed K,Mahfoud Z. Surface reactivity and self-oscillating oxidation of butan-2-ol over palladium loaded AlPO4[J].Appl Catal,A,2015,503:84-93.
[18] Li Meijun,Wu Zili,Overbury S H. Surface structure dependence of selective oxidation of ethanol on faceted CeO2nanocrystals[J].J Catal,2013,306(1):164-176.
[19] Gambaro L A,Briand L E. In situ quantification of the active acid sites of H6P2W18O62·nH2O heteropoly-acid through chemisorption and temperature programmed surface reaction of isopropanol[J].Appl Catal,A,2004,264(2):151-159.
[20] Mann A K P,Wu Zili,Calaza F C,et al. Adsorption and reaction of acetaldehyde on shape-controlled CeO2nanocrystals:Elucidation of structure-function relationships[J].ACS Catal,2014,4(8):2437-2448.
[21] Chen Dan,Qu Zhenping,Zhang Weiwei,et al. TPD and TPSR studies of formaldehyde adsorption and surface reaction activity over Ag/MCM-41 catalysts[J].Colloids Surf,A,2011,379(1/3):136-142.
[22] Molinari J E,Nakka L,Kim Taejin,et al. Dynamic surface structures and reactivity of vanadium-containing molybdophosphoric acid(H3+xPMo12-xVxO40) Keggin catalysts during methanol oxidation and dehydration[J].ACS Catal,2011,1(11):1536-1548.
[23] Nakka L,Molinari J E,Wachs I E. Surface and bulk aspects of mixed oxide catalytic nanoparticles:Oxidation and dehydration of CH3OH by polyoxometallates[J].J Am Chem Soc,2009,131(42):15544-15554.
[24] Wachs I E,Jehng Jih-Mirn,Ueda W. Determination of the chemical nature of active surface sites present on bulk mixed metal oxide catalysts[J].J Phys Chem B,2005,109(6):2275-2284.
[25] Guerrero-Pérez M O,Kim Taejin,Baňares M A,et al. Nature of catalytic active sites for Sb-V-O mixed metal oxides[J]. J Phys Chem C,2008,112(43):16858-16863.
[26] Routray K,Briand L E,Wachs I E. Is there a relationship between the M==O bond length(strength) of bulk mixed metal oxides and their catalytic activity?[J].J Catal,2008,256(1):145-153.
[27] Lee E L,Wachs I E. Surface chemistry and reactivity of welldefined multilayered supported M1Ox/M2Ox/SiO2catalysts[J]. J Catal,2008,258(1):103-110.
[28] Chen Dan,Qu Zhenping,Shen Shijin,et al. Comparative studies of silver based catalysts supported on different supports for the oxidation of formaldehyde[J].Catal Today,2011,175(1):338-345.
[29] Qu Zhenping,Shen Shijin,Chen Dan,et al. Highly active Ag/SBA-15 catalyst using post-grafting method for formaldehyde oxidation[J]. J Mol Catal A:Chem,2012,356:171-177.
(待续)
(编辑 王 萍)
Applications of temperature programmed surface reaction technique to the research for solid catalysts
Man Yi,Huang Wenqing,Chen Song
(Sinopec Beijing Research Institute of Chemical Industry,Beijing 100013,China)
The basic principles,equipments and experimental conditions of temperature programmed surface reaction(TPSR) technique were introduced. The applications of the TPSR technique in the research for catalysts in petrochemical industry in recent ten years were summarized in detail,which included the reforming oxidation of olefins and alkyls,Fischer-Tropsch synthesis,catalytic reduction of NOx,oxidation reduction of alcohol and aldehyde,oxidation of CO,and decomposition of compounds containing nitrogen,sulfur and chlorine. The development of TPSR in future was discussed.
temperature programmed surface reaction;solid catalyst;petrochemical industry;treatment of environmental pollutants
1000-8144(2017)03-0384-07
TQ 426.81
A
10.3969/j.issn.1000-8144.2017.03.020
2016-11-11;[修改稿日期]2016-12-30。
满毅(1983—),男,山东省青岛市人,硕士,高级工程师,电话 010-59202148,电邮 many.bjhy@sinopec.com。
