基于特征图谱的黄芩黄酮类成分固相萃取研究
2017-05-10郭威王亮周倩聂颖兰崔宁于宗渊
郭威,王亮,周倩, 聂颖兰, 崔宁 ,于宗渊*
(1. 山东中医药大学药学院,山东 济南 250355;2.山东省中医药研究院,山东 济南250014;3.中国中医科学院医学实验中心, 北京 100700)
【中药与天然活性产物】
基于特征图谱的黄芩黄酮类成分固相萃取研究
郭威1,2,王亮1,2,周倩1,2, 聂颖兰3, 崔宁2,于宗渊2*
(1. 山东中医药大学药学院,山东 济南 250355;2.山东省中医药研究院,山东 济南250014;3.中国中医科学院医学实验中心, 北京 100700)
采用HPLC法建立黄芩中黄酮类成分的特征图谱,以6个主要特征峰的回收率和特征图谱相似度为指标,优选固相萃取小柱填料、淋洗液和洗脱条件。最终确定C18固相萃取小柱对6个成分的回收率最高,淋洗液为甲醇-0.3%甲酸(20∶80,V/V),洗脱液为甲醇(含0.3%甲酸),纯化后6个主要特征峰的回收率均在89%以上,HPLC特征图谱相似度大于0.98。采用HPLC特征图谱指导优化黄芩黄酮类成分的固相萃取纯化方法,可保持纯化前后黄芩有效组分中化学成分的一致性。
黄芩;HPLC特征图谱;固相萃取;黄酮类成分
由于中药具有成分复杂多样且极性差异较大的特点,因此其有效组分的分离纯化一直是中药样品前处理过程中亟待解决的重要问题。固相萃取作为一种试样预处理技术,能有效地将分析物与干扰组分分离,具有高选择性、高回收率、低溶剂消耗、操作简便的特点[1-2],近年来将其应用于中药有效组分的富集和纯化中的研究日益增多。但在纯化条件的研究中,多以一种或几种有效成分的含量为考察指标,不符合中药多成分的特点。特征图谱作为一种综合评价中药质量的手段,将其作为指标优化纯化条件,能够准确地保证有效组分分离前后的一致性。
黄芩为唇形科植物黄芩(ScutellariabaicalensisGeorgi)的干燥根,有清热燥湿、泻火解毒、止血安胎等功效[3]。主要有效成分为黄酮类化合物,还含有少量油脂类、蜡质等弱极性物质以及氨基酸、糖等强极性物质,其理化性质有较大差异。本文采用固相萃取小柱来分离纯化黄芩中的黄酮类成分,除去弱极性及强极性的干扰物质,快速分离和富集目标成分,提高分析检测时的灵敏度,降低样品对分析柱的污染,并以特征图谱作为固相萃取分离纯化黄芩中黄酮类有效组分的评价手段,以特征图谱的相似度和几种主要有效成分的回收率为指标,综合探讨纯化条件,保证黄酮类成分的最大保留。
1 仪器与试药
1.1 仪器
1200系列高效液相色谱系统(美国安捷伦公司);6320离子阱质谱检测器(美国安捷伦公司),配备电喷雾离子源;Rotavapor R-3旋转蒸发仪(瑞士步琪公司);BP211D型电子天平(德国赛多利斯集团);Simplicity纯水仪(美国密理博公司);KDM型控温电热套(鄄城华鲁电热仪器有限公司)。
1.2 试药
AccuBondII SPE ODS-C18Cartridges固相萃取柱(美国安捷伦公司,产品编号188-1320);Cleanert PA 聚酰胺固相萃取小柱(天津博纳艾杰尔科技有限公司);高效液相色谱用甲醇为色谱纯,水为超纯水,甲酸等其他试剂均为分析纯。
黄芩为市售中药饮片,经山东省中医药研究院中药资源研究室林慧彬研究员鉴定,均为唇形科植物黄芩(ScutellariabaicalensisGeorgi)的干燥根。
2 方法与结果
2.1 黄芩提取液的制备
称取黄芩粉末(过三号筛)0.3 g,置具塞锥形瓶中,加60 mL水,称定重量,加热回流1 h,放冷,用水补足减失的重量,摇匀,过滤,即得供试品溶液1。
2.2 黄芩HPLC特征图谱的建立
2.2.1 色谱条件
色谱柱:Thermo Syncronis C18(250 mm×4.6 mm,5 μm)色谱柱;流动相:乙腈(A)-0.3%磷酸(B),梯度洗脱(0~30 min,85%~75%B;30~45 min,75%~70%B;45~50 min,70%~60%B;50~55 min,60%B;55~60 min,60%~40%B;60~65 min,40%~30%B;65~75min,30%~0%B);柱温:30 ℃;流速:1.0 mL/min;检测波长:280 nm;进样量:10 μL。色谱图见图1。

图1 黄芩提取液的HPLC特征图谱Fig.1 HPLC characteristic chromatogram of Radix Scutellariae extract
2.2.2 精密度实验
取同一批黄芩提取液,重复进样6次,按“2.2.1”项下色谱条件进行测定,记录特征图谱,6次测定图谱10个特征峰的相对保留时间的相对标准偏差均小于1%,相对峰面积的相对标准偏差均小于3%,仪器精密度良好。
2.2.3 重复性实验
取同一批黄芩按2. 1项下方法制备6份样品,分别测定,6份样品的10个特征峰相对保留时间的相对标准偏差小于1%,相对峰面积的相对标准偏差小于3%,表明方法的重复性良好。
2.2.4 稳定性实验
取新制备的黄芩提取液,分别于0、2、4、8、12、24 h进样,测定图谱的10个特征峰相对保留时间的相对标准偏差为0.07%~0.58%,相对峰面积的相对标准偏差为0.54%~2.91%,表明该方法处理的黄芩提取液在24 h内稳定。
2.2.5 黄芩HPLC特征图谱中特征峰的结构解析
根据DAD在线紫外光谱信息,初步判断1~10号峰为黄酮类化合物,结合文献,通过HPLC-MSn质谱信息对黄芩特征峰进行结构解析,结果见表1。

表1 黄芩HPLC特征图谱中特征峰的质谱解析
1号峰和2号峰的分子量相同,为同分异构体,推断可能为白杨素-葡萄糖-阿拉伯糖苷,且1、2号峰二级质谱碎片相同但碎片丰度不同,见图2。推测是由于阿拉伯糖存在呋喃型和吡喃型两种结构,六元环椅式构象比五元环折叠式稳定。因此,在相同质谱条件下,呋喃型阿拉伯糖基易于开环,消耗的碰撞能量少,从而易于产生较大的中性丢失;吡喃型阿拉伯糖基相对稳定,开环消耗的能量大,较小的中性丢失较多。由于六元环氧孤对电子易被分散,原子之间的电负性小,因此极性小,而五元呋喃糖的电子分散程度及结构稳定程度低于六元吡喃糖,各原子之间的电负性较大,所以呋喃糖极性大于吡喃糖,在反相柱中先流出。故推断1号峰为白杨素-葡萄糖-呋喃阿拉伯糖苷,2号为白杨素-葡萄糖-吡喃阿拉伯糖苷。
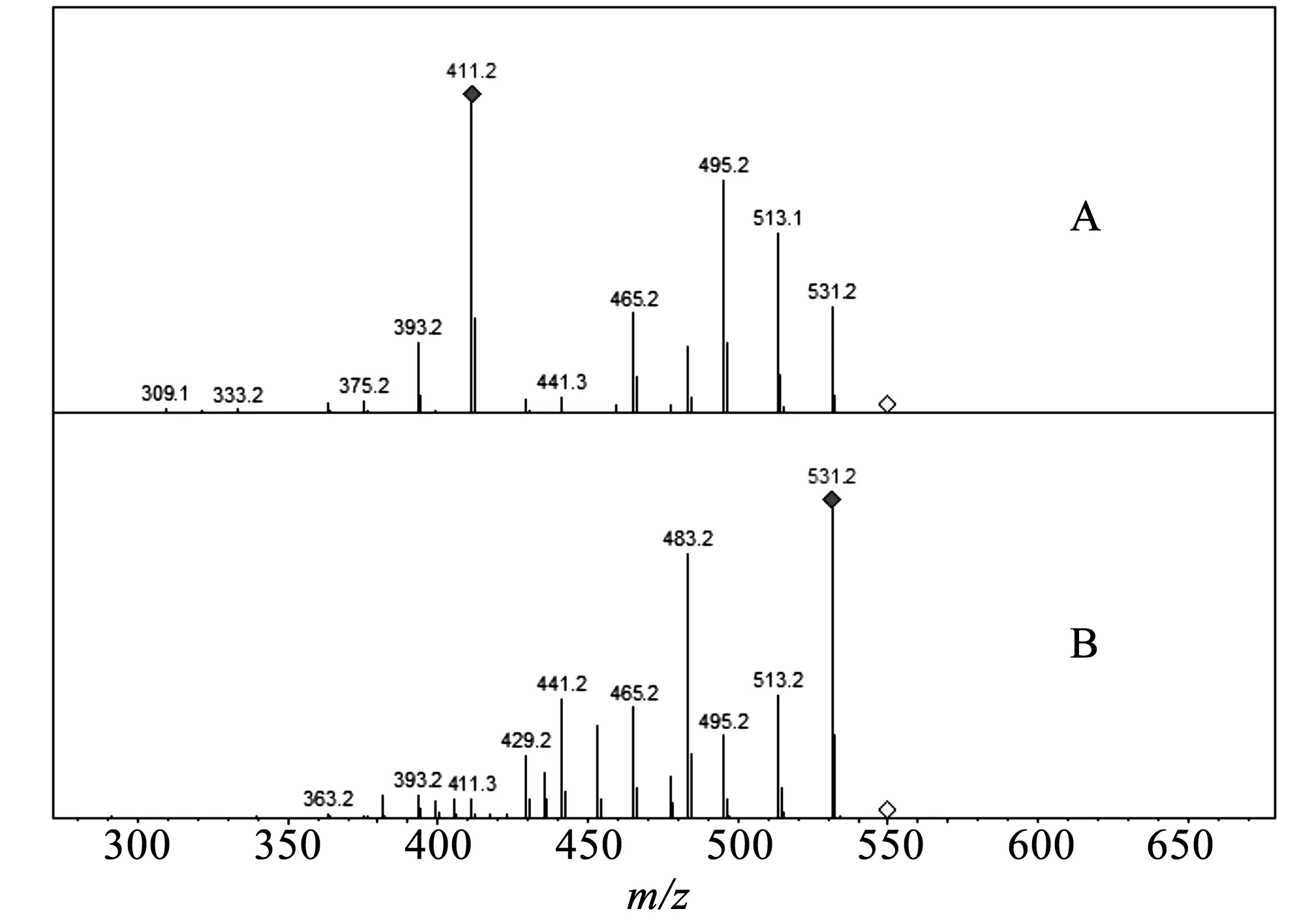
A 1号峰; B 2号峰。图2 1号峰和2号峰的二级质谱图Fig.2 Second-stage mass spectrum diagrams of peak 1 and peak 2
2.3 黄芩中黄酮类成分固相萃取纯化条件的优化
2.3.1 在不同洗脱剂下固相萃取小柱填料的影响
取不同类型的SPE小柱,分别用8 mL甲醇和8 mL水预淋洗。
精密量取按2.1项下方法制备的黄芩提取液1 mL,上样,用3 mL甲醇-0.3%甲酸(20∶80,V/V)分3次淋洗SPE小柱,每次1 mL,弃去淋洗液,吹出残留溶剂,用表2中的洗脱剂进行洗脱,收集洗脱液至50 mL梨形瓶中,旋转蒸发至干,加1 mL甲醇,振荡溶解,经0.45 μm滤膜滤过,即得供试品溶液2。
以供试液2的HPLC图谱中峰面积较大的6个特征成分(3,4,6,7,8,9号峰)为指标,与未经固相萃取处理的供试液1比较,计算各主峰的回收率[9]。
回收率(%)=(供试品溶液1主峰峰面积/供试品溶液2主峰峰面积)×100% 。
采用国家药典委员会出版的《中药色谱指纹图谱相似度评价系统2004A版》软件计算各洗脱液特征图谱与黄芩提取液指纹图谱相似度。
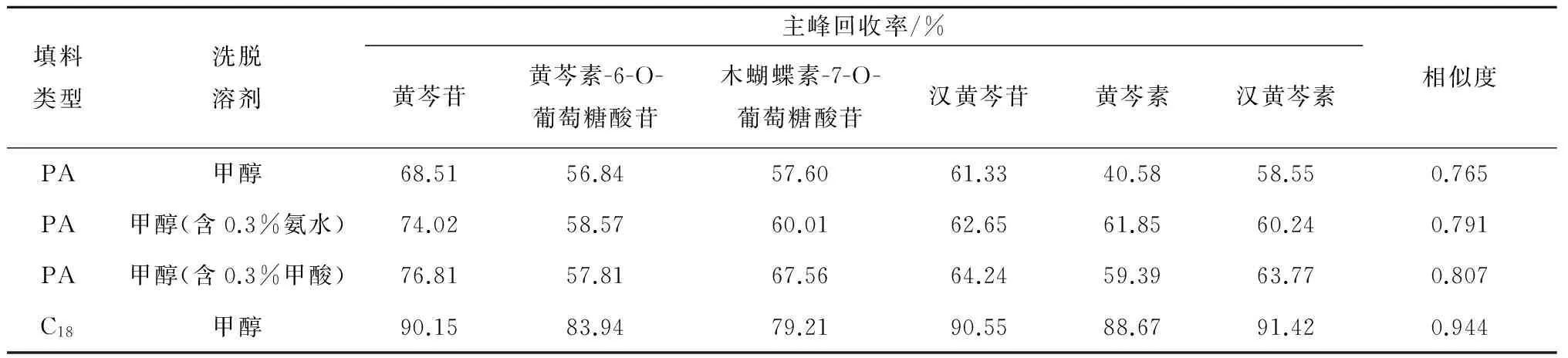
表2 填料类型对主峰回收率和特征图谱相似度的影响
表2的结果表明,在其他工艺条件都相同的情况下,不同填料类型的固相萃取小柱对黄芩提取液中各成分的保留率不同,当洗脱溶剂为甲醇时,聚酰胺小柱与C18小柱相比,各主峰保留率远远小于C18小柱;当洗脱溶剂为甲醇(含0.3%氨水)、甲醇(含0.3%甲酸)时,主峰保留率有所提高,但仍低于C18小柱,并且洗脱液的特征图谱与黄芩提取液特征图谱的相似度也均低于C18小柱,因此选择C18小柱。
2.3.2 淋洗液种类及洗脱液种类的考察
取1 mL黄芩提取液,上样,分别用水、甲醇-0.3%甲酸(20∶80,V/V)、甲醇-0.3%甲酸(40∶60,V/V)、甲醇-0.3%甲酸(60∶40,V/V)、甲醇-0.3%甲酸(80∶20,V/V)、甲醇、甲醇(含0.3%甲酸)分三次淋洗C18SPE小柱,每次1 mL,收集每个淋洗梯度的洗脱液至50 mL梨形瓶中,旋转蒸发至干,加1 mL甲醇,振荡溶解,按2.2.1中色谱条件测定3,4,6,7,8,9号峰面积。结果见表3。

表3 淋洗液及洗脱液种类对主峰回收率和特征图谱相似度的影响
水和甲醇-0.3%甲酸(20∶80,V/V)洗脱液中无特征成分出现,故选择甲醇-0.3%甲酸(20∶80,V/V)为淋洗液,既可将氨基酸、糖等强极性物质洗脱下来,又不影响特征成分的回收率。由表3结果可见,甲醇-0.3%甲酸(40∶60,V/V)洗脱液中开始出现特征成分,黄芩苷、黄芩素-6-O-葡萄糖酸苷、木蝴蝶素-7-O-葡萄糖酸苷、汉黄芩苷、黄芩素主要集中在甲醇-0.3%甲酸(60∶40,V/V)和甲醇-0.3%甲酸(80∶20,V/V)洗脱液中,甲醇-0.3%甲酸(60∶40,V/V)洗脱液与原提取液的相似度最高,但洗脱不完全。汉黄芩素主要集中于甲醇-0.3%甲酸(80∶20,V/V)洗脱液中,甲醇洗脱液中基本无特征成分,但当洗脱液换为甲醇(含0.3%甲酸)时,又可将部分特征成分洗脱下来,故确定以甲醇(含0.3%甲酸)为洗脱液。
2.3.2 洗脱液用量的考察
精密量取按2.1项下方法制备的黄芩提取液1 mL,上样,用3 mL甲醇-0.3%甲酸(20∶80,V/V)分3次淋洗C18SPE小柱,每次1 mL,弃去淋洗液,吹出残留溶剂,采用甲醇(含0.3%甲酸)洗脱,每次1 mL,共洗脱4次,收集洗脱液至50 mL梨形瓶中,旋转蒸发至干,加1 mL甲醇,振荡溶解,经0.45 μm滤膜滤过,按2.2.1中色谱条件测定,结果见图3。结果表明,第三次洗脱后仍有特征成分出现,第四次洗脱后无特征峰,即3 mL 的甲醇(含0.3%甲酸)可将吸附的黄酮类成分基本洗脱完全,因此确定洗脱液的用量为3 mL。

A第一次洗脱; B第二次洗脱; C第三次洗脱; D第四次洗脱。图3 不同体积甲醇洗脱液的色谱图Fig.3 Chromatograms of methanol eluent at different volume
2.3.6 方法专属性
精密吸取蒸馏水1 mL上样,用3 mL甲醇-0.3%甲酸(20∶80,V/V)分3次淋洗SPE小柱,每次1 mL,弃去淋洗液,吹出残留溶剂,用3 mL甲醇-0.3%甲酸(20∶80,V/V)洗脱,收集洗脱液至50 mL梨形瓶中,旋转蒸发至干,加1 mL甲醇,振荡溶解,经0.45 μm滤膜滤过,按2.2.1项下方法测定,得空白溶液的色谱图(见图4)。由空白与黄芩提取液的色谱图(图1)比较可知,用本实验拟定固相萃取方法处理黄芩提取液后,在70 min色谱记录时间内无干扰成分流出。

图4 空白溶液的色谱图Fig.4 Chromatogram of blank solution
2.3.7 验证实验
称取黄芩粉末(过三号筛)0.3 g,置具塞锥形瓶中,加60 mL 水,称定重量,加热回流1 h,放冷,用水补足减失的重量,摇匀,过滤,得黄芩提取液。取C18固相萃取小柱,分别用8 mL甲醇和8 mL水预淋洗SPE小柱。精密量取黄芩提取液1 mL,上样,用3 mL甲醇-0.3%甲酸(20∶80,V/V)分3次淋洗SPE小柱,每次1 mL,弃去淋洗液,吹出残留溶剂,用3 mL甲醇(含0.3%甲酸)洗脱,收集洗脱液至50 mL梨形瓶中,旋转蒸发至干,加1 mL甲醇,振荡溶解,经0.45 μm滤膜滤过,按2.2.1中色谱条件测定,计算各主峰的回收率(见表4)。结果表明,黄芩提取液经固相萃取处理后黄芩苷、黄芩素-6-O-葡萄糖酸苷、木蝴蝶素-7-O-葡萄糖酸苷、汉黄芩苷、黄芩素、汉黄芩素的回收率均在89%以上,重现性较好,说明确定的固相萃取小柱填料类型、淋洗液种类、洗脱液种类及用量等条件较为合理,重复性好且主成分保留率较高。黄芩中黄酮类成分的HPLC特征图谱与黄芩水提液的HPLC特征图谱相似度均大于0.98,实现了成分的均衡回收[9]。

表4 验证实验结果(n=3)
3 讨论
本实验采用HPLC特征图谱,对黄芩水提液中黄酮类成分的固相萃取条件进行了考察,结果表明用甲醇-0.3%甲酸(20∶80,V/V)淋洗,甲醇(含0.3%甲酸)洗脱,可以特异性地获取黄酮类成分,6个特征成分的回收率均在89%以上。在考察固相萃取淋洗和洗脱条件中发现,甲醇(含0.3%甲酸)洗脱能力强于纯甲醇,可能由于黄酮类成分多呈酸性,加酸后可抑制其电离,控制其呈分子状态,减少了洗脱时间,故加入甲酸后使甲醇的洗脱能力增强。
与固相萃取前相比,黄芩水提取物经固相萃取后浸膏重量减少了10%,故本实验建立的固相萃取方法可除去多糖等杂质,有利于减少非特征成分干扰、缩短分析时间和保护分析柱。
本实验以黄芩中6个特征成分(黄芩苷、黄芩素-6-O-葡萄糖酸苷、木蝴蝶素-7-O-葡萄糖酸苷、汉黄芩苷、黄芩素、汉黄芩素)的回收率和特征图谱的相似度作为评价指标,跟踪评价固相萃取的处理效果,与传统的以单一成分保留率为指标的评价方法相比更为精细,可较全面地反映固相萃取前处理中药样品时其药效物质的回收率,更好地体现了中药多成分的特征,可为中药中目标成分的分离与富集方法提供参考。
[1]赖宇红,陈浩桉.固相萃取法在中药分析中的应用前景及其建立优化新方法的策略与步骤[J].广东药学,2002,12(2):12-16.
[2]汤道权,高媛媛,魏雅芹,等.银杏叶提取物中四种黄酮类成分固相萃取技术优化[J].中药材,2008,31(6): 918-920.
[3]国家药典委员会.中华人民共和国药典2015年版一部[S].北京:中国医药科技出社,2015:301.
[4] OHKOSHI E,NAGASHIMA T,SATO H,et al. Simple preparation of baicalin from Scutellariae Radix[J]. J Chromatogr A, 2009, 1216(11): 2192-2194.
[5]SHANG X F,HE X R,HE X Y,et al.The genus Scutellaria an ethnopharmacological and phytochemical review[J]. J Ethnopharmacol,2010,128(2):279-313.
[6]HAN J,YE M,XU M,et al. Characterization of flavonoids in the traditional Chinese herbal medicine-Huangqin by liquid chromatography coupled with electrospray ionization mass spectrometry[J].J Chromatogr B Analyt Technol Biomed Life Sci,2007, 848(2):355-362.
[7] 赵胜男,李守拙.黄芩药材中黄酮类成分的HPLC-MS研究[J].承德医学院学报,2012, 29 (4):355-347.
[8] 杨思敏,陈瑞战,董航,等.高效液相色谱-质谱联用快速筛选并鉴定黄芩甲醇提取物中抗氧化活性成分[J].分析化学,2012,40(6):888-892.
[9] 杨红美,曾建国,陈波,等.具有指纹特征的黄芩提取物质量可控性研究[J].中草药,2006,37(6):846-851.
[10] 李守信,展金祥,刘武占,等.基于指纹图谱的金银花物质组纯化工艺研究[J].中草药,2015,46(1):55-60.
Solid phase extraction of flavonoids from Radix Scutellariae based on specific chromatogram
GUO Wei1,2, WANG Liang1,2, ZHOU Qian1,2,NIE Ying-lan3,CUI Ning2,YU Zong-yuan2*
(1. School of Pharmaceutical Sciences, Shandong University of Traditional Chinese Medicine, Jinan 250355, China;2. Shandong Academy of Chinese Medicine, Jinan 250014, China; 3. Experimental Research Center, China Academy of Chinese Medical Sciences, Beijing 100700, China)
∶The specific chromatogram of flavonoids in Radix Scutellariae was established by HPLC in this paper. The column filling, eluent and the elution condition of solid phase extraction were optimized using the recovery rates and specific spectral similarity of six main characteristic peaks in HPLC specific chromatogram as indexes. Experimental results confirmed that C18solid phase extraction (SPE) possessed the highest recovery rates of six components. The SPE column was rinsed with methanol-0.3% formic acid(20∶80,V/V), and then was eluted finally with methanol (containing 0.3% formic acid). After being purified, the recovery rates of six components were all over 89%, and the similarity of characteristic chromatogram was higher than 0.98. Based on HPLC characteristic chromatogram, the optimized purification process of flavonoids in Radix Scutellariae with solid phase extraction can ensure that the main effective components remain consistent before and after purification of Radix Scutellariae.
∶Radix Scutellariae; HPLC specific chromatogram; solid phase extraction; flavonoids
10.3976/j.issn.1002-4026.2017.02.003
2016-07-27
山东省中医药科技发展计划(2015-164)
郭威(1982—),女,硕士,助理研究员,研究方向为中药分析与质量控制。E-mail:331527800@qq.com
*通信作者,于宗渊,博士,研究员,研究方向为中药分析与质量控制。E-mail:yuzys@sohu.com
R284.2
A
1002-4026(2017)02-0013-07
