吸收光谱法测定不同价态钚的含量
2017-05-10伊小伟党海军林剑锋
伊小伟, 党海军, 曾 斌, 林剑锋
(西北核技术研究所, 西安 710024)
吸收光谱法测定不同价态钚的含量
伊小伟, 党海军*, 曾 斌, 林剑锋
(西北核技术研究所, 西安 710024)
用带有50 cm液芯波导毛细管池的光纤光谱仪测定了混合溶液中不同价态钚[Pu(Ⅲ)、Pu(Ⅳ)和Pu(Ⅵ)]的含量。根据Pu(Ⅲ)、Pu(Ⅳ)和Pu(Ⅵ)的吸收光谱特征,分别选择600,474,828 nm作为测定这3种离子的波长,并选择在其吸收曲线的波谷区640,492,850 nm依次作为参比波长。将在测定波长处所测吸光度减去在参比波长处的吸光度,从而得到上述3种价态钚离子的有效吸光度以扣除基线漂移等所导致的偏差。同时,用单一价态不同浓度的钚离子分别测定其在3个波长处的吸光度绘制标准曲线,并计算得其有效摩尔吸光率。将所测得的有效吸光度及有效摩尔吸光率代入相应的计算公式,即可计算得到上述3种价态钚离子的含量。本方法的测定结果与TTA萃取法所测得的结果比较,两种方法的测定结果在不确定度范围内是一致的。
吸收光谱法; 钚; 价态
钚是核燃料后处理流程中重点关注的元素之一。由于不同价态钚的氧化还原电位之间的差值较小,钚在硝酸溶液中常以多种价态共存,最常见的价态有Pu(Ⅲ)、Pu(Ⅳ)和Pu(Ⅵ)等3种[1-2]。无论在核燃料后处理普雷克斯(PUREX)流程中,还是在钚的放化分析中,都需要通过控制钚在硝酸溶液中的价态来实现分离纯化回收钚的目的,价态的稳定性和调价反应的动力学对流程的效果影响较大,这些通常都是通过经验的优化工艺来实现。为深入认识钚在硝酸溶液中的化学行为,需对钚在硝酸溶液中的价态分布进行研究。
目前,研究钚价态分布的方法有放射化学分析方法,主要利用萃取[3-6]、离子交换[7]和沉淀载带[8]或者多种方法联合等分离技术,快速将不同价态的钚分离,通过测定各组分中钚含量来确定钚的价态分布。化学方法的灵敏度高,但可能破坏已有的价态分布平衡,不能获得钚在溶液中真实的价态分布。激光诱导光声光谱法[9]和毛细管电泳法[10]也可用于钚的价态的确定,但操作系统复杂、仪器昂贵,难以用于现场应用。紫外-可见吸收光谱法可以根据不同价态钚离子的吸收光谱进行钚的价态的确定,直接获取钚在溶液中的价态分布,普通的分光光度计的检出限通常只能达到1×10-2~1×10-3mol·L-1,不能用于环境污染排放源等低浓度钚的价态的确定。
长光程液芯波导毛细管池的发展为分光光度法确定低浓度钚的价态提供了新的技术,长光程液芯波导毛细管池(LWCC)主要利用入射光在四氟乙烯的无定形聚合物与石英的交界面发生全反射,从而将光束束缚在波导内传播,在溶液量有限的条件下可有效增加吸收的光程。文献[11]报道利用50 cm长光程液芯波导毛细管池和光纤技术建立了高灵敏的钚价态确定装置,灵敏度比普通1 cm吸收池的分光光度计提高了45倍,但其工作主要聚焦钚价态确定装置研制,对光谱分析和最大吸收波长选择等方面未开展详细研究,对钚价态的确定是利用文献报道的单点波长处的摩尔吸光率进行计算。文献[12]利用1 m长光程液芯波导毛细管池与紫外-可见光谱仪对高氯酸体系中钚的4种氧化态进行确定,灵敏度比普通分光光度计提高了18~33倍,未开展钚在常用的硝酸体系中的价态分布研究。本工作利用配置50 cm液芯波导毛细管池的光谱仪开展钚在硝酸体系中价态分布的研究,建立了同时测定不同价态钚的方法,并通过萃取法和萃取色层法进行验证。该方法除满足实验室快速分析外,也可以作为一种快速、实时和在线的非介入技术,在生产工艺条件监控中发挥不可取代的作用。
1 原理
利用不同氧化态的吸收光谱确定溶液中钚的价态是广泛使用的价态确定方法,其中以Pu(Ⅲ)、Pu(Ⅳ)和Pu(Ⅵ)的价态为主。由于配制长光程液芯波导毛细管池的光谱仪是单光路单样品池,空白参比溶液的光谱与样品的光谱不是同时获取,仪器的基线漂移或波动不能有效扣除,同系列样品溶液的吸收光谱的基线不同,直接利用Pu(Ⅲ)、Pu(Ⅳ)和Pu(Ⅵ)在波长600,474,828 nm处的单点吸光度计算浓度存在偏差。一般可以选择其附近的谷区吸收带(这里分别为波长640,492,850 nm处)作为参比,以最大吸收波长和相应的参比吸收带吸光度之差作为该最大吸收波长处的有效吸光度,以减小由于基线漂移而导致的偏差。测定各种价态的钚离子在选用的多个吸收波长处的有效摩尔吸光率(ε),则混合价态溶液在每个波长处的吸光度等于每种价态的离子在该波长处的吸光度之和,见公式(1)~公式(3)。

2 试验部分
2.1 仪器与试剂
QE65PRO型光纤光谱仪,配50 cm液芯波导毛细管池。
钚(Ⅳ)标准储备溶液:1.43×10-3mol·L-1。
242Pu稀释剂:56.34 ng·g-1。
噻吩甲酰三氟丙酮(TTA)溶液:0.5 mol·L-1,以二甲苯为介质。
戊基磷酸二戊酯(UTEVA)树脂:取适量UTEVA树脂,用去离子水浸泡两次(每次浸泡24 h),滤去悬浮物,用3 mol·L-1硝酸溶液浸泡两次(每次浸泡24 h),滤去悬浮物,已处理好的树脂用3 mol·L-1硝酸溶液浸泡备用。
氨基磺酸亚铁溶液:0.4 mol·L-1,将氨基磺酸2.8 g溶于25 mL水中,加入还原铁粉0.56 g,加盖密封,待其自然溶解反应约3~4 d,基本澄清后即可认为反应完全。所配溶液需保存在棕色瓶中置于阴暗处,有效期为15~20 d。
硝酸、氨基磺酸、还原铁粉为优级纯,硝酸铈铵、噻吩甲酰三氟丙酮为分析纯,试验用水为超纯水。
2.2 试验方法
2.2.1 Pu(Ⅲ)、Pu(Ⅳ)和Pu(Ⅵ)摩尔吸光率的测定
称取不等量的1.43×10-3mol·L-1钚(Ⅳ)标准储备溶液至含有0.04 mol·L-1氨基磺酸亚铁的1 mol·L-1硝酸溶液中,反应约30 min后,Pu(Ⅳ)被氨基磺酸亚铁完全还原至Pu(Ⅲ),得到Pu(Ⅲ)的浓度在4×10-5~1.2×10-4mol·L-1内的多份溶液。以含有0.04 mol·L-1氨基磺酸亚铁的1 mol·L-1硝酸溶液为参比溶液,分别测量一系列Pu(Ⅲ)溶液的吸收光谱,根据已知的Pu(Ⅲ)浓度和溶液在波长474,600,828 nm处的吸光度绘制标准曲线。
称取不等量的1.43×10-3mol·L-1钚(Ⅳ)标准储备溶液至1 mol·L-1硝酸溶液中,得到Pu(Ⅳ)的浓度在4×10-5~10-4mol·L-1内的多份溶液。以1 mol·L-1硝酸溶液为参比溶液,分别测量一系列Pu(Ⅳ)溶液的吸收光谱,根据已知的Pu(Ⅳ)浓度和溶液在波长474,600,828 nm处的吸光度绘制标准曲线。
称取不等量的1.43×10-3mol·L-1钚(Ⅳ)标准储备溶液至含有0.05 mol·L-1硝酸铈铵的1 mol·L-1硝酸溶液中,反应约40 min后,Pu(Ⅳ)被硝酸铈铵完全氧化至Pu(Ⅵ),得到Pu(Ⅵ)的浓度在1×10-5~7×10-5mol·L-1内的多份溶液。以含有0.05 mol·L-1硝酸铈铵的1 mol·L-1硝酸溶液为参比溶液,分别测量一系列Pu(Ⅵ)溶液的吸收光谱,根据已知的Pu(Ⅵ)浓度和溶液在波长474,600,828 nm处的吸光度绘制标准曲线。
2.2.2 萃取法
称取一定量的1.43×10-3mol·L-1钚(Ⅳ)标准储备溶液于5 mL离心管中,加入一定量的含有0.001 mol·L-1硝酸铈铵的1 mol·L-1硝酸溶液,摇匀,放置60 min以确保氧化反应结束[使Pu(Ⅳ)被部分氧化]。反应完成后的溶液进行光谱测量,获得Pu(Ⅳ)和Pu(Ⅵ)的百分比。将测量完毕的溶液采用0.5 mol·L-1TTA-二甲苯溶液萃取两次,达到Pu(Ⅳ)和Pu(Ⅵ)分离的目的。完全分相后,称量有机相和水相的质量,从有机相[Pu(Ⅳ)]和水相[Pu(Ⅵ)]中分别称取一定量的溶液于15 mL塑料瓶中,加入56.34 ng·g-1242Pu稀释剂混匀,除去有机物,制源进行电感耦合等离子体质谱法(ICP-MS)测定,获得Pu(Ⅳ)和Pu(Ⅵ)的百分比。
2.2.3 萃取色层法
称取一定量的1.43×10-3mol·L-1钚(Ⅳ)标准储备溶液于5 mL离心管中,加入一定量的含有0.001 mol·L-1氨基磺酸亚铁的1 mol·L-1硝酸溶液,摇匀,放置60 min以确保还原反应结束[使Pu(Ⅳ)被部分还原]。反应完成后的溶液进行光谱测量,获得Pu(Ⅲ)和Pu(Ⅳ)的百分比。将部分测量结束的溶液上UTEVA树脂,依次采用1 mol·L-1硝酸溶液20 mL和含0.04 mol·L-1氨基磺酸亚铁的1 mol·L-1硝酸溶液20 mL将Pu(Ⅲ)和Pu(Ⅳ)分别洗脱。分别移取一定量的洗脱液于15 mL塑料瓶中,加入56.34 ng·g-1242Pu稀释剂,混匀,制源进行ICP-MS测定,获得Pu(Ⅲ)和Pu(Ⅳ)的百分比。
3 结果与讨论
3.1 Pu(Ⅲ)、Pu(Ⅳ)和Pu(Ⅵ)最大吸收波长的选择
试验扫描了不同浓度Pu(Ⅲ)的1 mol·L-1硝酸溶液的吸收光谱,典型的光谱见图1。

图1 Pu(Ⅲ)在1 mol·L-1硝酸溶液中的吸收光谱Fig. 1 Absorption spectrum of Pu(Ⅲ) in 1 mol·L-1 HNO3 solution
由图1可知:Pu(Ⅲ)在波长560,600,663 nm处均有吸收。由于Pu(Ⅳ)在吸收波长560,663 nm附近也存在吸收带,因此选择Pu(Ⅲ)的最大吸收波长为600 nm。为了减小由于基线漂移而导致的偏差,选择相应的参比吸收带为640 nm[13]。
1 mol·L-1硝酸溶液中Pu(Ⅳ)的典型吸收光谱见图2。

图2 Pu(Ⅳ)在1 mol·L-1硝酸溶液中的吸收光谱Fig. 2 Absorption spectrum of Pu(Ⅳ) in 1 mol·L-1 HNO3 solution
由图2可知:Pu(Ⅳ)在474,545,655 nm等多个波长处都有吸收。考虑Pu(Ⅲ)在吸收波长545,655 nm附近也存在吸收,因此选择Pu(Ⅳ)的最大吸收波长为474 nm,选择相应的参比吸收带为492 nm[13]。
Pu(Ⅵ)在1 mol·L-1硝酸溶液中的典型吸收光谱见图3。

图3 Pu(Ⅵ)在1 mol·L-1硝酸溶液中的吸收光谱Fig. 3 Absorption spectrum of Pu(Ⅵ) in 1 mol·L-1 HNO3 solution
由图3可知:Pu(Ⅵ)只在波长828 nm处有吸收,因此选择Pu(Ⅵ)的最大吸收波长为828 nm,选择相应的参比吸收带为850 nm[13]。
3.2 标准曲线
由Pu(Ⅲ)在波长474,600,828 nm处的标准曲线,拟合的方程分别见公式(4)~公式(6)。

由Pu(Ⅳ)在波长474,600,828 nm处的标准曲线,拟合的方程见公式(7)~公式(9)。
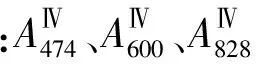


3.3 不同价态钚浓度的计算
将上述试验获得的一系列摩尔吸光率分别代入公式(1)~公式(3)中得到公式(12)~公式(14):


A828=256cⅢ-46cⅣ+1.92×104cⅥ
解方程,可计算出cⅢ、cⅣ、cⅥ见公式(15)~公式(17):
cⅢ=(1.54A474+4.91A600+0.019 6
cⅣ=(3.003A474-0.217A600+0.038 1
cⅥ=(1.059-0.5
由此,可以得到多种价态共存条件下各种价态钚的浓度。
3.4 光谱分析方法的检验
TTA萃取剂只萃取Pu(Ⅳ),不萃取Pu(Ⅵ),可采用TTA将Pu(Ⅳ)和Pu(Ⅵ)分离,然后分别测定有机相和水相中Pu(Ⅳ)和Pu(Ⅵ)的浓度,计算Pu(Ⅳ)和Pu(Ⅵ)的价态分布。采用光谱法和TTA萃取法获得同一溶液中Pu(Ⅳ)和Pu(Ⅵ)的百分比,结果见表1。

表1 Pu(Ⅳ)和Pu(Ⅵ)的测定结果Tab. 1 Determination results of Pu(Ⅳ) and Pu(Ⅵ)
由表1可知:两种方法获得Pu(Ⅳ)和Pu(Ⅵ)分布的结果在不确定度范围内一致。
UTEVA树脂只吸附Pu(Ⅳ),不吸附Pu(Ⅲ),可采用UTEVA萃取色层法将Pu(Ⅲ)和Pu(Ⅳ)分离,然后分别测定Pu(Ⅲ)和Pu(Ⅳ)的浓度,计算Pu(Ⅳ)和Pu(Ⅲ)的价态分布。采用光谱法和UTEVA萃取色层法获得同一溶液中Pu(Ⅳ)和Pu(Ⅲ)的百分比,结果见表2。

表2 Pu(Ⅳ)和Pu(Ⅲ)的测定结果Tab. 2 Determination results of Pu(Ⅳ) and Pu(Ⅲ)
由表2可知:两种方法获得Pu(Ⅳ)和Pu(Ⅲ)分布的结果在不确定度范围内一致。
本方法可以推广到不同浓度硝酸溶液中钚的价态确定,需要根据不同价态的钚在特定浓度硝酸溶液中的吸收光谱特征选择相应的最大吸收波长和参比吸收带,并绘制相应的标准曲线。
[1] CHOPPIN G R. Humics and radionuclide migration[J]. Radiochimica Acta, 1988,44/45:23-28.
[2] SOOD D D, PATIL S K. Chemistry of nuclear fuel reprocessing: current status[J]. J Radioanal Nucl Chem, 1996,203(2):547-573.
[3] FOTI S C, FREILING E C. The determination of the oxidation states of tracer uranium, neptunium and plutonium in aqueous media[J]. Talanta, 1964,11:385-392.
[4] NITSCHE H, LEE S C, GATTI R C. Determination of plutonium oxidation states at trace levels pertinent to nuclear waste disposal[J]. J Radioanal Nucl Chem, 1988,124(1):171-185.
[5] BERTRAND P A, CHOPPIN G R. Separation of actinides in different oxidation states by solvent extraction[J]. Radiochim Acta, 1982,31:135-137.
[6] SAITO A, CHOPPIN G R. Separation of actinides in different oxidation states from neutral solutions by solvent extraction[J]. Anal Chem, 1983,55:2454-2457.
[7] GEHMECKER H, TRANTMANN N, HERRMANN G. Separation of plutonium oxidation states by ion exchange chromatography[J]. Radiochim Acta, 1986,40:81-88.
[8] CLEVELAND J M, REES T F, NASH K L. Plutonium speciation in selected basalt, granite, shale, and tuff groundwaters[J]. Nucl Technol, 1983,62:298-310.
[9] NEU M P, HOFFMAN D C. Comparison of chemical extractions and laser photoacoustic spectroscopy for the determination of plutonium species in near-neutral carbonate solutions[J]. Radiochimica Acta, 1994,66/67:251-258.
[10] KUCZEWSKI B, MARQUARDT C M, SEIBERT A, et al. Separation of plutonium and neptunium species by capillary electrophoresis-inductively coupled plasma-mass spectrometry and application to natural groundwater samples[J]. Anal Chem, 2003,75:6769-6774.
[11] 付建丽,吴继宗,张丽华,等.高灵敏钚价态分析仪的研制[J].核化学与放射化学, 2013,35(1):8-13.
[12] WILSON R E, HU Y J, NITSCHE H. Detection and quantification of Pu(Ⅲ,Ⅳ,Ⅴ,Ⅵ) using a 1.0-meter liquid core waveguide[J]. Radiochim Acta, 2005,93:203-206.
[13] MAHILDOSS D J, RAVI T N. Spectrophotometric determination of plutonium Ⅲ, Ⅳ, and Ⅵ concentrations in nitric acid solution[J]. J Radioanal Nucl Chem, 2012,294:87-91.
Absorption Spectrophotometric Determination of Plutonium in Different Oxidation States
YI Xiao-wei, DANG Hai-jun*, ZENG Bin, LIN Jian-feng
(NorthwestInstituteofNuclearTechnology,Xi′an710024,China)
Optical fiber absorption spectrophotometer equipped with 50 cm liquid core waveguide capillary cell was used for determination of plutonium in various oxidation states, i.e. Pu(Ⅲ), Pu(Ⅳ) and Pu(Ⅵ), in a mixed solution. Based on the spectrometric characteristic of Pu in the 3 oxidation states, 600, 474, 828 nm were chosen as the wavelengths for measuring the absorbances of Pu(Ⅲ), Pu(Ⅳ) and Pu(Ⅵ) respectively. To eliminate deviations due to drift of baselines in measurements of absorbances, values of absorbances were measured at the reference wavelengths at 640, 492, 850 nm, which were selected from the trough of the respective measuring wavelengths, and substracted from their values of absorbance of Pu(Ⅲ), Pu(Ⅳ) and Pu(Ⅵ) respectively, to obtain their valid values of absorbance. Simultaneously, standard curves and valid values of molar absorptivities were determined with Pu standard solutions of single oxidation state by measuring the absorbances at the related wavelengths. The values obtained were substituted into the given formulas and contents of Pu(Ⅲ), Pu(Ⅳ) and Pu(Ⅵ) in the mixture were calculated. The results obtained were checked by the TTA extraction method, and consistency within the range of uncertainty between the results of the 2 methods was attained.
Absorption spectrophotometry; Plutonium; Oxidation states
10.11973/lhjy-hx201702008
2016-07-11
科技部科技基础性工作专项(2015FY110800)
伊小伟(1981-),男,湖北随州人,副研究员,主要研 究方向为光谱分析。
* 通信联系人。E-mail:danghaijun@nint.ac.cn
O657.31
A
1001-4020(2017)02-0160-05
