肾病综合征、低钾、代谢性碱中毒
2017-05-09张志宏徐维玮王金泉
张志宏 徐维玮 王金泉
·临床集锦·
肾病综合征、低钾、代谢性碱中毒
张志宏 徐维玮 王金泉
64岁女性,四肢乏力4年,水肿1周入院。临床表现大量蛋白尿,低白蛋白血症,伴低钾、低镁血症,代谢性碱中毒,低尿钙;血浆肾素活性增高,高醛固酮血症;肾活检病理提示肾小球足细胞病变,一处可疑肾小球旁器肥大。基因筛查显示COL4A5基因突变(823C>G)伴UMOD基因突变(1653G>C),而SLC12A1、KCNJ1、CLCNKB和BSND及SLC12A3基因均未见异常。综合患者临床表现与实验室检查最后诊断为肾小球足细胞病伴失盐性肾小管疾病。
肾病综合征 失盐性肾小管疾病 UMOD基因 COL4A5基因
病史摘要
现病史 64岁女性,因“四肢乏力4年,水肿1周”入院。4年前患者无明显诱因劳动时出现四肢无力,休息后可缓解,不伴肌肉痉挛,未诊治。1周前发现双下肢水肿,伴尿泡沫增多。2015年10月至我科门诊,查尿蛋白3+,血清白蛋白(Alb)28.2 g/L,血钾2.7 mmol/L,肝酶及血清肌酐(SCr)正常范围,为进一步诊治入院。病程中无肉眼血尿,无脱发、眼干口干、烦渴多饮,无皮疹、关节痛。纳差,长期素食,无慢性呕吐、腹泻。
既往史及个人史 无高血压、糖尿病病史,无利尿剂、缓泄剂用药史,无吸烟、饮酒史,否认家族性肾脏病史与近亲婚配。
体格检查 体温36.8℃,脉搏80次/min,呼吸16次/min,血压98/70 mmHg,身高145 cm,体重33.5 kg,未见龋齿,无口角糜烂、舌乳头萎缩,浅表淋巴结未及肿大,心肺腹部未见特殊;双下肢轻度水肿。
实验室检查
尿液 尿蛋白定量3.54 g/24h,尿沉渣红细胞计数5万/ml(多形型),白细胞0~1/HP、管型阴性;RBP 11.1 mg/L,Cystatin C 0.89 mg/L;NGAL 79.7 μg/L,IL-18 49.55 ng/ml;尿渗量554 Osm/(kg·H2O),尿单克隆游离轻链κ 210.0 mg/L,λ 100.9 mg/L,κ/λ 2.1。一次性尿液检查:钠133.9 mmol/L,钾48.4 mmol/L,钙0.98 mmol/L,尿磷8.69 mmol/L,尿肌酐4 839 μmol/L,尿葡萄糖7.92 mmol/L,尿酸2 108 μmol/L,尿氯116 mmol/L。24h尿液生化:尿钠121 mmol/24h,尿钾119 mmol/24h,尿氯186 mmol/24h,尿钙2.3 mmol/24h。滤过钾排泄分数(FEK%)31.1%,跨肾小管钾离子梯度(TTKG)9.05,尿钾与尿肌酐比值(uK/Cr) 89.6 mmol/g,滤过氯排泄分数(FECl%)2.1%。
血液
血常规 Hb 121 g/L,WBC 5.9×109/L,N/L 67.7/21.8%,PLT 216×109/L。
血生化 Alb 24.2 g/L,Glo 26.2 g/L,AST 22 U/L,ALT 19 U/L;BUN 17.3 mg/dL,SCr 84 μmol/L,UA 155 μmol/L;Chol 6.40 mmol/L,TG 2.38 mmol/L,HDL-ch 0.53 mmol/L,LDL-ch 2.16 mmol/L;钾2.7 mmol/L,钠131 mmol/L,镁0.6 mmol/L,钙1.94 mmol/L,磷0.82 mmol/L,氯92.3 mmol/L,TCO229.8 mmol/L,血糖5.86 mmol/L,糖化血红蛋白5.7%。eGFR(CKD-EPI公式) 63.05 ml/(min·1.73m2)。
免疫学 IgG 8.95 g/L,IgA 2.1 g/L,IgM 0.654 g/L,ASO 32 IU/ml,RF<20 IU/ml;补体C31.0 g/L,C40.2 g/L;ANA、抗ds-DNA阴性,抗核抗体谱阴性。外周血淋巴细胞亚群:CD3 672个/μl,CD4 429个/μl,CD8 201个/μl,CD20 127个/μl,Treg 22个/μl。抗磷脂酶A2受体抗体阴性。血清单克隆游离轻链κ 46.9 mg/L,λ 25.9 mg/L,免疫固定电泳图谱κIgG深染但无条带聚集。
肿瘤标志物 甲胎蛋白、癌胚抗原、β2微球蛋白、铁蛋白与肿瘤相关抗原CA19-9、CA125、CA15-3、CA24-2均正常范围。

肾素-血管紧张素-醛固酮系统(RASS) (立位)血浆肾素268.53 pg/ml,血浆肾素活性13.44 ng/ml·h(参考值0.93~6.56 ng/ml·h),血管紧张素Ⅱ 317.00 pg/ml,醛固酮357.92 pg/ml(参考值40~310 pg/ml);(卧位)血浆肾素64.41 pg/ml,血浆肾素活性 5.63 ng/ml·h(参考值0.05~0.79 ng/ml·h),血管紧张素Ⅱ 195.24 pg/ml,醛固酮266.46 pg/ml(参考值10~160 pg/ml)。
辅助检查
双肾超声 左肾108 mm×42 mm×54 mm,右肾99 mm×46 mm×49 mm,未见肾盂肾盏扩张。肝胆胰脾超声均未见异常。心脏超声:二尖瓣及主动脉瓣钙化。甲状腺超声:右叶囊实性团块,TI-RADS 超声分级3级。胸片:左侧胸膜增厚钙化。心电图:窦性心律,RV5+SV1>4.0 mV。
滤纸条试验泪液浸湿长度>10 mm,泪膜破裂时间>10s。听力测验及眼部裂隙灯检查正常。
肾活检病理
光镜 42个肾小球中1个球性废弃,余肾小球节段外周袢足细胞胞质少。系膜区增宽不明显,毛细血管袢开放好,一处可疑球旁器肥大。灶性肾小管上皮细胞刷状缘脱落,偶见小管上皮细胞等立方空泡变性。偶见小管萎缩、基膜增厚,管腔内见蛋白管型,间质散在单个核细胞浸润,并小灶性分布,动脉未见病变(图1、2)。

图1 肾小球毛细血管袢开放好,系膜区增宽不明显(Masson三色,×400)

图2 可疑肾小球旁器肥大(↑)(Masson三色,×400)
免疫荧光 肾小球、肾小管、管间毛细血管及间质血管均未见免疫复合物和补体沉积。κ、λ轻链染色阴性。
电镜 肾小球足细胞病变明显,足细胞足突融合约50%~60%,少量微绒毛化。肾小球系膜区略增宽,系膜区未见电子致密物沉积。肾小球毛细血管袢开放好,偶见内皮下疏松、区域增宽,血管袢基膜内皮下、上皮侧未见电子致密物沉积,肾小球基膜(GBM)厚约260~440 nm,数处基膜与基膜紧贴(图3)。
小结:肾小球足细胞病变明显,节段基膜与基膜紧贴。

图3 肾小球足细胞足突融合,系膜区未见电子致密物沉积(EM)
诊疗分析
患者临床特点:(1)老年女性,起病隐匿;(2)肾脏受累表现双下肢水肿,大量蛋白尿、低蛋白血症,高脂血症;低钾低镁血症,代谢性碱中毒,尿钙减少。根据上述特点,入院初步诊断为肾病综合征。
导致成人肾病综合征的原发性肾脏疾病病理类型主要有膜性肾病、足细胞病。既往我科对13 519例肾活检资料的分析显示膜性肾病(MN)占原发性肾小球肾炎的9.89%[1]。MN 发病年龄以>40岁多见,血清磷脂酶A2 受体抗体阳性对诊断具有重要指示作用[2],免疫病理IgG沿GBM呈弥漫颗粒状沉积是MN 特征性病变。足细胞病是依肾脏病理形态学特征命名的一组疾病范畴,足细胞为受损的靶细胞[3],微小病变肾病(MCD)和局灶节段性肾小球硬化(FSGS)是其中典型代表。肾活检病理肾小球基本正常,免疫病理肾小球系膜区和毛细血管袢无免疫复合物沉积,电镜下见足细胞广泛的足突融合,而GBM厚度和结构正常。本例患者肾活检免疫荧光染色阴性,光镜下肾小球病变轻微,电镜下肾小球足细胞广泛足突融合脱落,GBM厚度和结构正常,“肾小球足细胞病”诊断成立。
本例特殊之处在于肾病综合征合并明显电解质及酸碱平衡紊乱,即低钾、低镁血症、代谢性碱中毒和低尿钙。结合患者病史与血液尿液检验结果,高度考虑合并Gitelman综合征(Gitelman syndrome,GS)。GS为常染色体隐性遗传性肾小管疾病,发病机制是位于染色体长臂(16q13)的SLC12A3基因突变引起,该基因编码噻嗪类利尿剂敏感钠氯共同转运子(NCCT)。NCCT功能缺陷可导致远曲小管重吸收NaCl障碍,肾性失钠失氯引起血容量减少,反馈激活RASS,醛固酮敏感的钠通道开放增加钠离子重吸收,促进钾离子和氢离子分泌,导致低血钾和代谢性碱中毒[4-5]。该患者在低血钾状态下,尿钾排泄量并未减少,符合肾性失钾[6-8];患者血压临界偏低,RASS检查提示高肾素活性和继发性高醛固酮血症,支持“GS”诊断。但需要除外以下疾病:(1)肾动脉狭窄:临床表现为中重度高血压,四肢血压不对称,腹部听诊闻及血管杂音等,肾动脉彩色多普勒超声是常用筛查手段;(2)原发性醛固酮增多症:临床表现为高血压伴低血钾,RASS检查显示血和尿醛固酮增高且不被抑制,血浆肾素活性降低:(3)Liddle综合征:是一种少见的常染色体显性遗传病,以高血压、低血钾、代谢性碱中毒、低血浆肾素和低醛固酮血症为特征。此外,也需要与假性Bartter综合征(BS)、GS或类GS相鉴别[9],后者常见病因有利尿剂或缓泻剂使用不当、长期低氯饮食、周期性呕吐、囊性纤维化与先天性失氯性腹泻。排除利尿剂使用,类GS时钾离子随尿液、粪便或呕吐物丢失,而肾小管无病变,尿氯排泄量均降低;而GS尿氯排泄量增高。本例患者尿钠与尿氯不低,但尿钙减低,更支持“GS”的诊断。
本例患者父母非近亲婚配,无肾脏病家族史,肾活检病理仅发现一处可疑肾小球旁器肥大。为明确诊断,对患者及其子女进行遗传性肾脏病相关基因筛查,结果显示患者COL4A5基因突变(823C>G)伴UMOD基因突变(1653G>C),而SLC12A1、KCNJ1、CLCNKB和BSND及SLC12A3基因均未见异常(表1)。
综上考虑,最后诊断肾小球足细胞病,失盐性肾小管疾病。治疗上嘱患者正常饮食,予泼尼松30 mg/d、螺内酯120 mg/d、氯化钾缓释片2.0 g/d、门冬酸钾镁0.84 g/d。每周复查血电解质。6周后门诊复诊,患者乏力症状缓解,尿蛋白定量0.43 g/24h,血Alb 33.2 g/L,钾4.15 mmol/L、镁2+1.23 mmol/L,肾功能稳定,予激素逐渐减量,随访观察中。
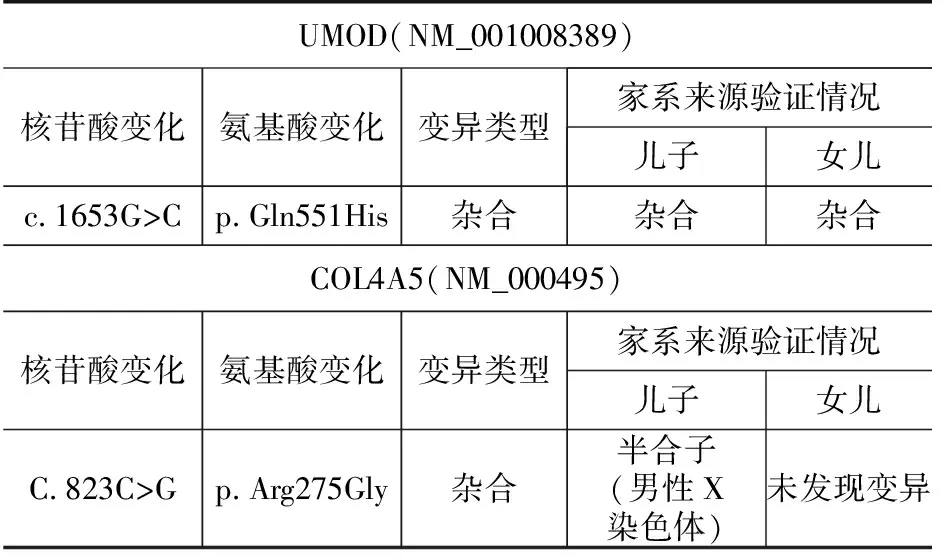
表1 患者及子女基因筛查结果
讨 论
本病例存在两点特殊性:(1)患者电解质与酸碱平衡紊乱高度怀疑归因于遗传性肾小管疾病,但基因筛查结果与临床推断不符;(2)遗传性肾小管疾病罕见合并肾病综合征,肾小球足细胞病变与基因突变内在联系尚未见文献报道。
根据患者临床表现与血尿检验结果,首先考虑诊断GS。GS通常20岁以后发病,症状相对较轻,表现为间歇性疲乏,肌无力,也可合并软骨钙质沉着。与GS相近的另一类遗传性肾小管疾病是BS,共分为Ⅰ~Ⅴ型,相关基因分别为SLC12A1(Ⅰ型);KCNJ1(Ⅱ型);CLCNKB(Ⅲ型);BSND(Ⅳ型);CASR(Ⅴ型)。临床对于GS、BS和类Gitelman的鉴别诊断有时较为困难。新近有研究纳入基因筛查确诊的30例BS(Ⅲ型)、90例GS和43例类Gitelman综合征患者,比较三组间临床与血尿生化结果的差异。结果发现三组间年龄、血清镁水平、尿钙与尿肌酐比值、血浆肾素活性以及醛固酮浓度指标上差异均显著,且类Gitelman综合征的FENa%与FECl%均最低[10]。有研究认为口服氢氯噻嗪试验(HCT试验)有助于三者鉴别。该研究纳入基因筛查确诊的GS 41例(其中儿童19例),BS 7例(Ⅰ型5例,Ⅲ型2例),类Gitelman综合征3例(利尿剂使用不当2例,慢性呕吐1例)。患者禁食12h后进行水化处理(饮水10 ml/kg)并持续卧床4h,水化后30 min给予HCT 50 mg(儿童1 mg/kg)顿服,每30 min收集一次尿液共6组,分别于第1h与第4h采集静脉血,计算服用HCT后最高FECl%与基础FECl%的差值。结果38例GS患者表现为“HCT试验反应迟钝”,即FECl%差值<2.3%,而BS及类Gitelman综合征患者均无该现象出现[10]。需要指出的是,HCT试验固然有助于鉴别诊断,但操作程序繁琐,对于严重低钾患者进行水化和利尿处理需慎重。
一般认为病理上见肾小球旁器肥大提示GS诊断,但也有基因筛查确诊的GS病例不伴有肾小球旁器肥大[11]。遗传性肾小管疾病确诊需要确定突变基因。本例对遗传性肾脏病有关的基因进行筛查,结果显示与BS发病有关的SLC12A1、KCNJ1、CLCNKB和BSND基因以及与GS发病有关的SLC12A3基因均未见异常。文献报道已发现的SLC12A3基因突变,包括错义突变、剪切突变、无义突变、读码框位移突变,其中大部分以错义突变为主,复合杂合突变多为纯合子突变,无热点突变[12]。本例采用的Bestseq靶向捕获测序技术基于高通量测序,不仅检测已知致病基因,还同时检测数百个相关可能的致病基因。本例未能发现SLC12A3基因突变,尚不明确是否存在不适合该技术检测的未知点突变类型或染色体平衡易位位点。
本例患者基因筛查意外发现COL4A5基因突变伴UMOD基因突变。一方面,COL4A5基因发生单个碱基错义突变,编码第275号氨基酸由精氨酸(Arg)变为甘氨酸(Gly)。现已明确,COL4A5基因突变与X连锁显性遗传型Alport综合征有关[13]。Alport综合征以肾小球源性血尿为主要临床表现,肾活检病理电镜下GBM广泛增厚伴致密层分裂为其典型病变,应用特异性的抗Ⅳ型胶原不同α链的抗体进行免疫荧光检查,GBMα3与α5链节段缺失。本例患者虽检出COL4A5基因突变,但缺乏肾病家族史和血尿现病史,眼耳检查未见异常,肾组织Ⅳ型胶原染色与电镜超微结构均未观察到Alport综合征典型表现,故不支持Alport综合征诊断。但新近发现COL4A5基因与成人FSGS样病变之间存在某种关联。采用新一代靶向测序技术对来自76个家庭(其中24个家庭有肾病家族史)的81名FSGS样病变成人患者进行Ⅳ型胶原相关基因筛查,结果在6个家庭8名成员中检测出合并COL4A 3~5基因突变[14]。另一方面,本例患者UMOD基因也发生单个碱基错义突变,编码第551号氨基酸由谷氨酰胺(Gln)变为组氨酸(His)。UMOD基因突变与常染色体显性遗传小管间质性肾病-尿调节素型(ADTKD-UMOD)[15-17]发病有关。UMOD基因定位于16p.12.3,正常情况下调控髓袢升支粗段小管上皮细胞编码Tamm-Horsfall蛋白(也称尿调节素)。UMOD基因错义突变可导致折叠错误的蛋白前体在内质网积聚,也可干扰Na+-K+-2Cl-共转运体向腔面膜转移,从而引起该段小管电解质重吸收和尿液浓缩障碍,继而反馈性增加近端小管电解质与尿酸重吸收。患者典型临床表现为高尿酸血症、痛风和肾功能不全,肾活检病理提示间质纤维化、小管萎缩,常规免疫荧光检查阴性,以特异性抗UMOD抗体染色在小管升支粗段上皮细胞可见折叠错误的Tamm-Horsfall蛋白积聚。本例患者实验室检查肾功能基本正常,无高尿酸血症,无肾脏病家族史,穿刺肾组织间质无明显纤维化,不支持ADTKD-UMOD诊断。
本文报告病例肾活检病理明确存在肾小球足细胞病变,经激素诱导后蛋白尿缓解,但COL4A5基因与GBM,UMOD基因与肾小管之间的内在联系,不能完全否认基因突变在其中的影响。既往观点认为经典的失盐性肾小管病变不会累及肾小球,与肾小球源性蛋白尿无关[18]。然而,从肾脏病理角度已观察到足细胞病变与GS共存,涉及到局灶节段性肾小球硬化(FSGS)[19-20]与C1q肾病[21]。通过敲除SLC12A3基因的小鼠模型,也观察到GBM出现节段性增厚,内在机制推测为RASS激活与低钾血症对足细胞的损伤作用[19]。
遗憾的是我们未能获得患者父母的遗传信息,对患者基因突变遗传背景无法准确把握。患者子女血液及尿液化验均未见异常,但其子X染色体COL4A5基因错义突变,子女UMOD基因均杂合突变,需要随访观察。
小结:本文报告1例老年女性患者,临床表现肾病综合征伴低钾、低镁血症和代谢性碱中毒,尿钙减少,血浆肾素活性增高伴继发性高醛固酮血症;肾活检病理提示肾小球足细胞病变伴一处可疑肾小球旁器增生;基因筛查显示UMOD与COL4A5基因错义突变,最终诊断为肾小球足细胞病,失盐性肾小管病变。经激素、纠正电解质紊乱等治疗,病情缓解。本例充分反映了遗传性肾小管疾病的遗传变异性与表观差异性,值得临床关注。
1 Li LS,Liu ZH.Epidemiologic data of renal diseases from a single unit in China:analysis based on 13,519 renal biopsies.Kidney Int,2004,66(3):920-923.
2 Ramachandran R,Kumar V,Kumar A,et al.PLA2R antibodies,glomerular PLA2R deposits and variations in PLA2R1 and HLA-DQA1 genes in primary membranous nephropathy in South Asians.Nephrol Dial Transplant,2016,31(9):1486-1493.
3 Bierzynska A,Soderquest K,Koziell A.Genes and podocytes-new insights into mechanisms of podocytopathy.Front Endocrinol (Lausanne),2015,5:226.
4 Knoers NV,Levtchenko EN.Gitelman syndrome.Orphanet J Rare Dis,2008,3:22.
5 Graziani G,Fedeli C,Moroni L,et al.Gitelman syndrome:pathophysiological and clinical aspects.QJM,2010,103(10):741-748.
6 Assadi F.Diagnosis of hypokalemia:a problem-solving approach to clinical cases.Iran J Kidney Dis,2008,2(3):115-122.
7 Choi MJ,Ziyadeh FN.The utility of the transtubular potassium gradient in the evaluation of hyperkalemia.J Am Soc Nephrol,2008,19(3):424-426.
8 Lin SH,Halperin ML,Hypokalemia:a practical approach to diagnosis and its genetic basis.Curr Med Chem,2007,14(14):1551-1565.
9 Matsunoshita N,Nozu K,Shono A,et al.Differential diagnosis of Bartter syndrome,Gitelman syndrome,and pseudo-Bartter/Gitelman syndrome based on clinical characteristics.Genet Med,2016,18(2):180-188.
10 Colussi G,Bettinelli A,Tedeschi S,et al.A thiazide test for the diagnosis of renal tubular hypokalemic disorders.Clin J Am Soc Nephrol,2007,2(3):454-460.
11 秦岭,邵乐平,任红,等.中国人Gitelman综合征高发突变的基因型和表型特征.肾脏病与透析肾移植杂志,2008,17(4):331-334.
12 Urbanová M1,Reiterová J,Stěkrová J,et al.DNA Analysis of Renal Electrolyte Transporter Genes Among Patients Suffering from Bartter and Gitelman Syndromes-Summary of Mutation Screening.Folia Biologica (Praha),2011,57:65-73.
13 Barker DF,Hostikka SL,Zhou J,et al.Identification of mutations in theCOL4A5 collagen gene in Alport syndrome.Science,1990,248(4960):1224-1227.
14 Gast C,Pengelly RJ,Lyon M,et al.Collagen (COL4A) mutations are the most frequent mutations underlying adult focal segmental glomerulosclerosis.Nephrol Dial Transplant,2016,31(6):961-970.
15 Ekici AB,Hackenbeck T,Morinière V,et al.Renal fibrosis is the common feature of autosomal dominant tubulointerstitial kidney diseases caused by mutations in mucin 1 or uromodulin.Kidney Int,2014,86(3):589-599.
16 Troyanov S,Delmas-Frenette C,Bollée G,et al.Clinical,Genetic,and Urinary Factors Associated with Uromodulin Excretion.Clin J Am Soc Nephrol,2016,11(1):62-69.
17 Eckardt KU,Alper SL,Antignac C,et al.Autosomal dominant tubulointerstitial kidney disease:diagnosis,classification,and management--A KDIGO consensus report.Kidney Int.2015,88(46):76-83.
18 Devuyst O,Konrad M,Jeunemaitre X.Tubular disorders of electrolyte regulation.In:Avner ED,Harmon WE,Niaudet P,eds.Pediatric Nephrology,6th ed.New York:Springer,2009,929-978.
19 Demoulin N,Aydin S,Cosyns JP,et al.Gitelman syndrome and glomerular proteinuria:a link between loss of sodium-chloride cotransporter and podocyte dysfunction? Nephrol Dial Transplant,2014,29(Suppl 4:):iv117-120.
20 Bulucu F,Vural A,Yenicesu M,et al.Association of Gitelman’s syndrome and focal glomerulosclerosis.Nephron,1998,79(2):244.
21 Hanevold C,Mian A,Dalton R.C1q nephropathy in association with Gitelman syndrome:a case report.Pediatr Nephrol,2006,21(12):1904-1908.
(本文编辑 莫 非 律 舟)
Nephrotic syndrome with hypokalaemia and metabolic alkalosis
ZHANGZhihong,XUWeiwei,WANGJinquan
NationalClinicalResearchCenterofKidneyDisease,JinlingHospital,NanjingUniversitySchoolofMedicine,Nanjing210016,China
A 64-year-old woman was referred to our hospital due to muscular weakness for four years and episodes of edema for one week. Laboratory tests showed macroalbuminuria, hypoalbuminemia, hypokalaemia, hypomagnesaemia, hypocalciuria, metabolic alkalosis, hyperreninemia, and hyperaldosteronism. Her renal biopsy revealed podocytopathy with hyperplasia of the juxtaglomerular apparatus. Genetic testing was performed and heterozygous mutations were found in UMOD and COL4A5 gene rather than SLC12A1, KCNJ1, CLCNKB, BSND and SLC12A3 gene. The final diagnosis for this patient was podocytopathy and salt-losing tubulopathy. We discuss the association of mutation in the UMOD and COL4A5 gene with glomerular proteinuria and salt-losing tubulopathies.
nephrotic syndrome salt-losing tubulopathy UMOD gene COL4A5 gene
10.3969/cndt.j.issn.1006-298X.2017.02.020
南京军区南京总医院肾脏科 国家肾脏疾病临床医学研究中心 全军肾脏病研究所(南京,210016)
2016-03-24
