Abnormal expression of TFIIIB subunits and RNA Pol III genes is associated with hepatocellular carcinoma☆
2017-05-07JunxiaLeiSonglinChenShupingZhong
Junxia Lei,Songlin Chen,Shuping Zhong
aSchool of Medicine,South China University of Technology,China
bDepartment of Cardiothoracic Surgery,Xiamen University Af fi liated Southeast Hospital,China
cDepartment of Biochemistry and Molecular Biology,Keck School of Medicine,University of Southern California,Los Angeles,CA,USA
1.Introduction
RNA polymerase III(Pol III)mediates the synthesis of avariety of small untranslated RNAs,including 5S rRNA,tRNAs,7SL RNA,U6 RNA,and Alu-associated microRNAs.1These noncoding RNAs are divided into 3 types:5S rRNA is Type 1,tRNAs are Type 2,and U6 RNA is Type 3.2
The products of Pol III-dependent genes(Pol III genes),such as tRNAs and 5S rRNA,are elevated in transformed and tumor cells,suggesting that they are crucial to tumorigenesis.The upregulation of Pol III genes enhances the capacity of cells to synthesize proteins,which is required for cell growth,proliferation,transformation,and tumor development.3-6
The promoters of tRNA genes require the transcription factor complexes TFIIIB and TFIIIC,in addition to Pol III(Fig.1A),for accurate and ef ficient transcription,whereas the 5S rRNA transcriptional machinery is composed of Pol III,TFIIIA,TFIIIB,and TFIIIC(Fig.1B).The TFIIIB complex contains three subunits:TATA boxbinding protein(TBP),Bdp1,and TFIIB-related factor 1(Brf1)or Brf2.The TFIIIC complexcomprises the TFIIIC63,TFIIIC 90,andTFIIIC 102 subunits in mammalian cells.Brf1 participates in the regulation of Type 1(5S rRNA)and Type 2(tRNAs)gene transcription,whereas Brf2 modulates that of Type 3 genes,such as U6.Studies have indicated that alterations in the activity of the TFIIIB complex are associated with cell transformation and tumorigenesis.3-6
TBP is required for transcription by all three nuclear polymerases.TBP combines with TAFs to form at least three distinct complexes-SL1,TFIID,and TFIIIB-which then specify its function in RNA Pol I,II,and III transcription,respectively.7In contrast,Brf1 and Bdp1 are used specifically in Pol III gene transcription.Changes in TBP expression alter the cellular levels of Bdp1 but not Brf1,8implying that Brf1 is more specific to and critical in the regulation of tRNA and 5S rRNA genes.
2.Dysregulation of Pol III genes promotes cell transformation and hepatocellular carcinoma(HCC)
2.1.Oncogenic proteins and tumor suppressors modulate TFIIIB activity to affect Pol III gene transcription
Oncogenic proteins,such as Ras,c-Jun,and c-Myc,stimulate Pol III gene transcription,5-7,9,10whereas the tumor suppressors BRCA1,pRb,p53,PTEN,and Maf1 repress transcription of this class of genes.5,6,9-13The ability of these oncogenic and tumor suppressor proteins to regulate Pol III gene transcription is attributed to their capacity to control the TFIIIB complex.
Modulation of TFIIIB activity is central in regulating Pol III gene transcription.The tumor suppressors RB and p53 bind directly to TFIIIB and prevent its recruitment to the promoters of Pol III genes.14-16PTEN negatively regulates Brf1 phosphorylation and its ability to associate with TBP to form functional TFIIIB complexes.12Bdp1 is phosphorylated during mitosis,inducing its selective dissociation from chromatin.17,18Maf1 suppresses Pol III gene transcription by repressing TBP expression and inhibiting TFIIIB recruitment to promoters.19BRCA1 de ficiency increases alcoholinduced Pol III gene transcription,whereas restoration of BRCA1 in cells mitigates this effect.11In contrast,cellular and viral oncogenic proteins induce Pol III gene transcription through enhanced expression of individual transcriptional components.
Hepatitis B virus(HBV)is an important cause of the development of HCC in Asia.The HBV oncogenic X protein increases TBP expression,requiring the activation of Ras.20Bdp1 expression is induced in cells that are transformed bypapovaviruses,21whereas a rise in Brf1 mRNA is observed in response to the integration of high-risk humanpapillomavirus.22TBP is upregulated in a clinically signi ficant number of human colon cancers.23Overexpression of Brf1 transforms mouse embryo fibroblasts and are in cells of cervical cancers and in biopsies of human HCC cases.24-26Our recent studies also indicate that Brf1 is overexpressed in estrogen receptor-positive(ER+)human breast cancer cases(unpublished).
Alcohol-mediated increases in TBP and Brf1 drive alcoholinduced liver tumor formation.6We have demonstrated that Brf1 is overexpressed and tRNA and 5S rRNA transcription is enhanced in human liver cancer patients.25Notably,these HCC cases with high Brf1 expression have shorter survivals,25suggesting that elevated Brf1 levels re flect the oncogenic status of cells and implicating it as a novel biomarker for HCC.
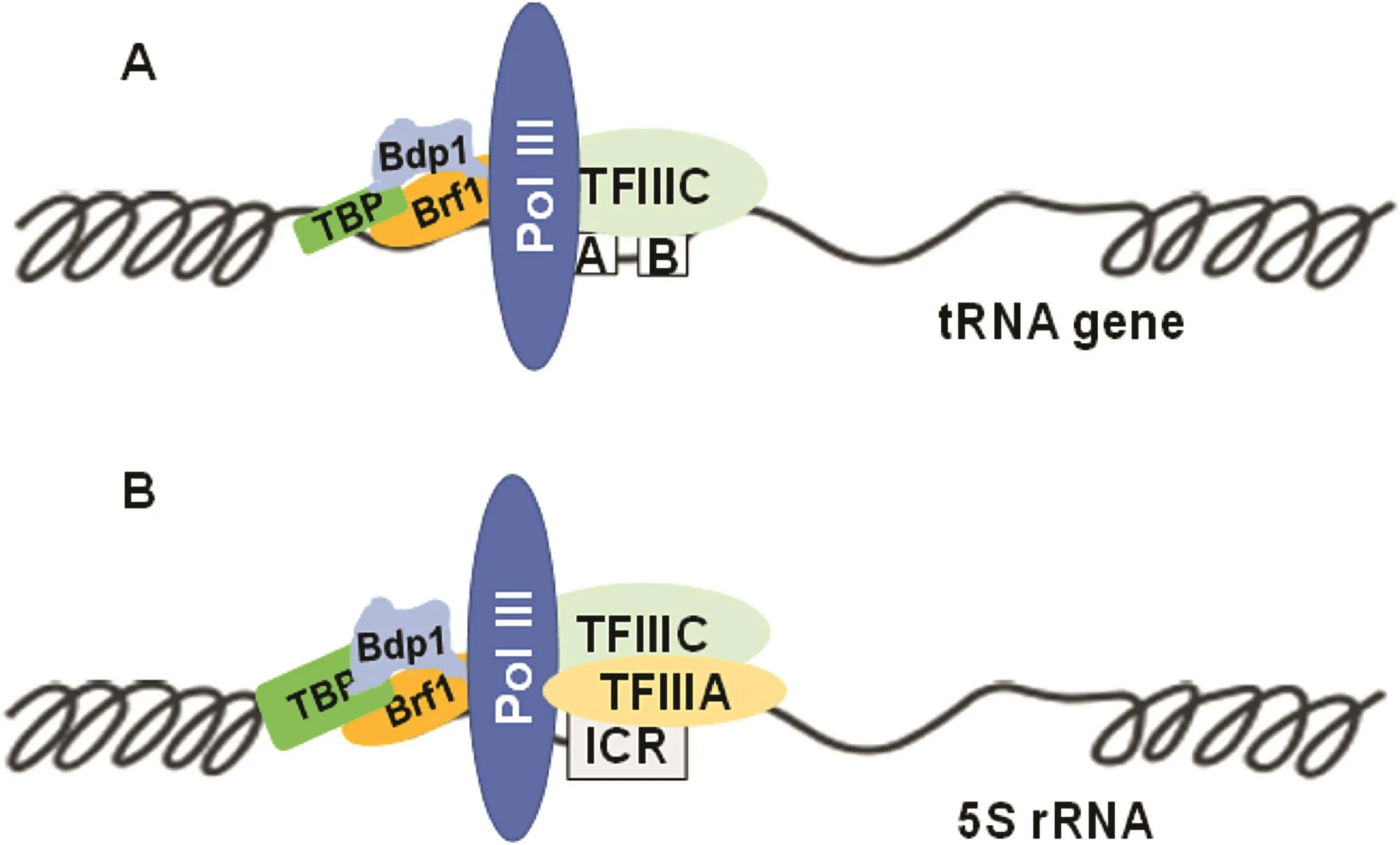
Fig.1.TFIIIB subunits are recruited to the promoter of Pol III genes.(A)Transcriptional machinery of tRNA genes includes Pol III,TFIIIC,and TFIIIB in the regulatory region of tRNAs.TFIIIB subunits(Brf1,TBP,Bdp1)interact with oncogenic proteins and suppressors.(B)Transcriptional machinery of 5S rRNA has Pol III,TFIIIA,TFIIIC,and TFIIIB in the regulatory region of 5S rRNA.A,B,and ICR are the regulatoryelements in the promoter regions of tRNA and 5S rRNA genes.tRNAs and 5S rRNA,the concentrations of which increase in transformed cells and tumor tissues,have important roles in protein synthesis.Abbreviations:Pol III,RNA polymerase III;TBP,TATA box-binding protein;Brf1,TFIIB-related factor 1.
2.2.Dysregulation of Pol III genes promotes cell transformation and tumor development
Neoplastic cells have increased levels of 5S rRNAs and tRNAs,and a high rate of Pol III gene transcription is a general feature of transformed and tumor cells;3,4,6,9,25,27-29thus,regulation of the synthesis of tRNAs and 5S rRNAs is a fundamental determinant of the oncogenic potential of cells.Pol III genetranscriptional products are crucial in tumorigenesis,24,25,27,28,30and enhanced Pol III gene transcription is required for oncogenic transformation.25-31A decrease in Brf1 reduces Pol III gene transcription,which is suf ficient for inhibiting cell transformation and tumor formation in nude mouse.3-6,27,28
On consuming alcohol,transgenic animals that express HCV NS5A protein,which present with more advanced fatty liver disease compared with wild-type mice,32show greater Pol III gene activity and upregulate TBP and Brf1 versus wild-type animals.In NS5A transgenic mice that form liver tumors,32Pol III gene activity is enhanced further.6A mechanistic analysis indicates that alcohol increases the cellular levels of c-Jun,which occupies the TBP and Brf1 promoters to enhance TBP and Brf1 expression and Pol III gene transcription.6Cases of human HCC with alcohol consumption have higher levels of Brf1 and Pol III gene expression,25indicating that alcohol-induced upregulation of these species is critical during the development of HCC.
Alcohol is considered to be carcinogenic in humans by the International Agency for Research on Cancer(IARC).33,34Target sites for alcohol-related carcinogenesis in humans include the liver and breast.
It is unknown whether there is a common mechanism through which alcohol-associated cancer develops in various human organs.Cytologically,cancer cells consistently show nucleolar hypertrophy,wherein RNA Pol III genes are transcribed.10This feature might allow us to identify a shared mechanism of alcoholassociated human cancers by examining the dysregulation of Brf1 and Pol III genes.3,4,6,25Animal experiments have shown that alcohol intake promotes tumor development.35,36Alcohol increases the expression of Brf1 and Pol III genes in liver and breast cell lines,and the cellular levels of Brf1 in cancer cell lines are higher than in nontumor lines.3-6,10,25,28,30,37Thus,alcoholinduced dysregulation of Brf1 and Pol III genes might constitute a common pathway of alcohol-associated cancers,29supporting that the induction of Pol III genes contributes to alcohol-associated liver tumor development.
3.Signaling events during dysregulation of Pol III genes
3.1.The JNK1 pathway is involved in liver tumor development
As discussed,the dysregulation of Brf1 and Pol III genes is critical in cell transformation and tumorigenesis.Thus,identifying their signaling pathways is essential for determining the molecular mechanism of the development of HCC.Diethylnitrosamine(DEN),a potent chemical hepatocarcinogen,has been used widely to induce HCC in rodents,causing HCC in 100%of male mice.38DEN disrupts the nuclear enzymes that are involved in DNA repair/replication and cancers.39Mice that lack JNK1 are much less susceptible to DEN-induced hepatocarcinogenesis.40Impaired tumorigenesisdue to JNK1 de ficiency correlateswith decreased expression of cyclin D and vascular endothelial growth factor(VEGF),diminished cell proliferation,and reduced tumor neovascularization.40Whereas hepatocyte-specific deletion of IKK augments DEN-induced hepatocyte death and cytokine-driven compensatory proliferation,disruption of JNK1 abrogates this response.40,41This compensatory proliferation of hepatocytes requires an increase in protein synthesis,which relies on elevated Brf1 expression and Pol III gene transcription,suggesting that the inhibition of Brf1 and Pol III gene activities suppresses the development of HCC.
3.2.JNK1 positively and JNK2 negatively mediate the expression of TFIIIB subunits and Pol III genes
JNKs are a class of mitogen-activated protein kinases(MAPKs),which are activated by stress,proin flammatory stimuli,and mitogenic factors.42,43JNKs are encodedbythree genes-JNK1,JNK2,andJNK3-which give rise to multiple isoforms that are generated by alternative splicing.44-46JNK1 and JNK2 are ubiquitously expressed,whereas JNK3 expression is restricted tothe brain,heart,and testis.46JNK1 and JNK2 possess structural and biochemical similarities and thus have many overlapping functions.JNKs induce apoptosis and enhance cell proliferation and transformation,depending on cell type.47,48
All three subunits of TFIIIB are modulated by JNKs.49The ability of JNKs to differentially control Poll III gene transcription is mediated through altered expression of the TFIIIB complex.JNK-mediated regulation of all 3 TFIIIB subunits is orchestrated through Elk-1.Although the function of Elk-1 in human cancer has not been clearly established,initial results indicate that downregulation of Elk-1 suppresses the tumorigenicity of human HCC cells.50Although Bdp1 expression is not regulated directly by Elk-1,the resulting Elk-1-mediated change in TBP expression governs Bdp1 promoter activity and cellular Bdp1 concentrations,demonstrating that alterations in cellular TBP levels modulate Bdp1 expression but not Brf1.8
JNK1 and JNK2 have disparate substrate specificities and gene targets.51-54Functions that are unique to JNK1 include its selective interaction with activating transcription factor 2(ATF2),54regulation of microtubule-associated proteins,51and ability to induce apoptosis in response totumor necrosis factoralpha.53The function of these JNKs in tumor development remains controversial.55,56Depending on the cell type and stimulus,JNKs stimulate oncogenic transformation and act as tumor suppressors.49
JNKs are important in regulating the stability and activity of certain transcriptional activators.Once JNKs become activated,they phosphorylate a variety of cellular targets,including transcription factors,such as c-Jun,JunD,ATF2,and Elk-1,resulting in their stimulation and subsequent alterations in gene activity.57In the absence of external stimuli,however,JNKs enhance the degradation of their substrates.58
We have demonstrated novel functions of JNKs in regulating the expression of TBP.TBP is a central transcriptional initiation factor,and changes in its levels have pleiotropic but selective effects on transcription by all three nuclear RNA polymerases.Increased TBP expression induces the transcription of genes that encode tRNAs and 5SrRNAs,5,20,59-61whereasprotein-encodinggenesare differentially regulated.62-64Because TBP levels in fluence the activities of a wide array of genes,JNK-mediated regulation of cellular TBP expression is likely to be a key mechanism by which JNKs impact cellular gene expression.Consistent with this model,we have found that JNKs control Pol III gene activity.49
3.3.Elk-1 binds toTBP and Brf1 promoter to mediate its transcription
JNK1 and JNK2 have opposing functions in regulating TBP expression.49The selective activation of JNK1 but not JNK2 is responsible for inducing TBP mRNA and protein expression in mouse and human cell lines.49Furthermore,JNK2 negatively regulates TBP expression in nonstimulated cells,consistent with previous studies that have demonstrated that JNK2 but not JNK1 undergoes autophosphorylation in the absence of upstreamactivated kinases.65TBP expression is induced by the activation of select signaling molecules and is negatively regulated by JNK2.49These studies support that the balance between JNK1 and JNK2 is an important mechanism that is responsible for maintaining the exquisite dynamic equilibrium in cellular TBP levels.
Because small increases inTBP promote transformation of rat 1A cells without changing proliferation rates,23,66the cell typedependent phenotypic effects of JNKs might result from their ability to regulate TBP expression.These studies imply that the balance between JNK1 and JNK2 is a key mechanism by which cellular TBP levels are regulated.49The opposing functions of JNK1 and JNK2 in governing TBP levels suggest that JNK1-mediated increases in TBP promote oncogenesis,whereas JNK2-mediated declines in TBP suppress oncogenesis.49The chief function of JNK1 and JNK2 is to regulate TBP levels,which ultimatelyenhances Pol III gene transcription and the growth potential and transformation of cells.
JNK1 and JNK2 mediate their effects on TBP expression through transcriptional regulation of the TBP promoter using a common Elk-1 binding site.49Elk-1 does not bind appreciably to this site in nonstimulated cells,but on EGF stimulation,Elk-1 is recruited to the promoter,correlating with its induction.49Interestingly,JNKs have antagonistic functions in regulating Elk-1 phosphorylation and Elk-1 binding to the TBP promoter.49Consistent with these results,JNK1 can directly phosphorylate Elk-1,67,68and increased phosphorylation of Elk-1 increases its DNA binding activity and its binding to the coactivator p300.69,70Elk-1 also directly regulates the Brf1 promoter,synchronizing the expression of these proteins.Accordingly,the Brf1 promoter contains a consensus Ets binding site at-671,near the transcriptional start site(TSS).Because Brf1 expression rises in cardiomyocytes when MEK1,an activatorof ERK,is overexpressed,71it is likely that the activation of multiple signaling pathways converge on Elk-1 to regulate Brf1 expression.However,we can not rule out the possibility that other JNK-targeted transcription factors control the Brf1 promoter and Pol III gene transcription.
Surprisingly,in contrast to previous studies that have demonstrated that JNK2 phosphorylates Elk-1 directlyin vitro,72cells that are de ficient for JNK2 have greater phosphorylated Elk-1 levels,which are augmented on EGF stimulation.This result shows that JNK2 does not phosphorylate Elk-1in vivoand that it negatively regulates the phosphorylation of Elk-1.Elk-1 is a direct substrate of all three classes of MAPKs.73It is conceivable that JNK2 specifically inhibits the ability of JNK1,p38,and ERK1/2 to phosphorylate Elk-1.Alternatively,JNK2 might prevent a priming kinase from phosphorylating Elk-1,thereby decreasing subsequent phosphorylation of Elk-1 by the other MAPKs.
However,that JNK2 decreases the phosphorylation of Elk-1 in nonstressed cells suggests that even in the absence of MAPK activation,JNK2 can still lower Elk-1 phosphorylation.Protein phosphatase2B negatively regulatesElk-1 byantagonizing the phosphorylation that is induced by MAPK activation.74Therefore,it is also plausible that JNK2 facilitates the dephosphorylation of Elk-1 through its ability to boost the function of an Elk-1-targeted phosphatase.In either case,these studies support a novel function of JNK2 in negatively regulating the phosphorylation and function of Elk-1.49
3.4.JNK1 and JNK2 differentially modulate the expression of TFIIIB subunits,TBP,and Brf1
Through chemical genetic methods that selectively inhibit the function of JNK2 in MEFs,the use of knockout cells has been suggested to be insuf ficient to derive conclusions with regard to the function of JNK2.75This study proposes an alternative explanation for the opposing functions of JNKs in MEFs and shows that in contrast to previous studies and the current studies,JNK2 positively,rather than negatively,regulates c-Jun expression and cellular proliferation,based on the finding that long-term loss of gene function affects an adaptive response and changes in the functions of other genes and a compensatory increase in JNK1 function inJnk2-/-cells.54,76
However,we used an additional approach and another cell line to examine the functions of JNK1 and JNK2.Transient repression of JNK2 expression in Huh-7 cells by approximately 4-fold by siRNA forless than 2 days had functional consequences onTBP expression,similar to those that were observed inJnk2-/-MEFs.Interestingly,treatment ofJnk2-/-MEFs with a chemical inhibitor of JNK2 was conducted within a similar time frame.75This argues against the notionthat the functional differences of JNK2 in MEFs are due tothe long-term effects of gene disruption.Nevertheless,further work is needed to determine how long-term loss of gene function affects the expression and function of other genes and the time that is required for these potential compensatory changes in genes to occur.
Our studies have identi fied a new mechanism by which JNKs regulate cellular concentrations of c-Jun-through their ability to governTBP expression.49c-Jun is regulated by JNKs,77and although JNK1 and JNK2 bind to and phosphorylate c-Jun,leading to its activation,JNKs target c-Jun for ubiquitination and degradation.78JNK1 increases c-Jun activity and stability,whereas JNK2 binds preferentially to c-Jun in nonstimulated cells and directs it toward degradation.53,54The tumor suppressor p16INK4a,which interacts selectively with JNK1 and JNK3,prevents c-Jun phosphorylation.79Thus,JNKs regulate c-Jun levels and activity through several mechanisms.
Although initialin vitrostudies have identi fied common JNK substrates,such as c-Jun,p53,and Elk-1,subsequent reports have demonstrated that JNK1 and JNK2 have opposing functions with regard to the stability and phosphorylation of these substratesin vivo.8,49Both JNKs phosphorylate c-Jun,but JNK1 positively regulates c-Jun stability,whereas JNK2 negatively regulates it.54Another substrate of these kinases,p53,is differentially regulated by JNKs.80Inhibition of JNK1 induces p53 expression,whereas that of JNK2 represses it in primary human fibroblasts.80Whether JNK2 antagonizes JNK1 or indirectly mediates the dephosphorylation of Elk-1 or other transcription factors remains to be determined.
The ability of JNKs to differentially regulate the function or expression of transcription factor substrates has potential pleiotropic effects on cellular gene expression.The opposing activities of JNKson Elk-1 phosphorylation simultaneouslycontrolsthe expression of TBP and its associated factors,Bdp1 and Brf1,which ultimately dictates the transcription of Pol III genes.TBP is a limiting component of Pol III gene transcription in Huh-7 cells and MEFs.3,4,8,49,81JNK alone alters the cellular levels of TBP,which can modulate Pol III gene transcription in these cells.JNK-mediated changes in cellular TBP expression control cell proliferation rates.49Furthermore,these changes regulate c-Jun levels and its activity,which in turn increase proliferation rates.50The regulation of Pol III gene transcription by JNKs through JNK effects on TBP and TBP-associated factors,Brf1 and Bdp1,constitutes a mechanism by which JNKs control the proliferative capacity of cells.
Although JNK targets are continuously being de fined,the analysis of the functions of JNKs in cellular growth responses has proven to be more complex.Animal studies have begun to examine such functions in cancer.Paradoxically,disruption ofJNK1in mice inhibits the transformation of pre-B cells by Bcr-Abl,82and these mice are more susceptible to phorbol ester-induced skin tumors than wild-type animals.83In contrast,JNK2-/-mice have a lower propensityfordevelopingphorbolester-induced papillomas,compared with wild-type mice.84Because the differences in the functions of JNKs are likely to be substrate-and cellular contextdependent,identifying JNK1-and JNK2-specific targets is crucial for understanding how they elicit distinct cellular responses.Further studies will be needed to determine the context-dependent functions of JNK isoforms in suppressing or inducing oncogenicity.
In addition to upregulating the TFIIIB subunit TBP,JNK1 increases Brf1 expression in liver and breast cells.4,6,25JNKs modulate Pol III gene transcription through Brf1.6,8Zhong and colleagues6,8have reported opposing functions of JNKs in regulating Pol III gene transcription.As the major products of the class of genes,tRNAs and 5S rRNAs are essential components of the protein synthesis machinery.Our studies have identi fied a link between stressinduced JNKs and translational capacity.JNK1 induces Pol III genes,whereas JNK2 negatively regulates transcription.JNK1 and JNK2 govern Pol III gene transcription-JNK1 positively regulates these genes following stress induction by anisomycin or alcohol,whereas JNK2 negatively regulates them in nonstimulated cells.6,8JNK2 also impairs Pol I transcription.85
Translational control is as an important step in oncogenic transformation,consistent with the transcription of these genes being tightly controlled by tumor suppressors and oncogenic proteins.5-13,86A causative role for Pol III gene transcription in determiningtheoncogenicstatusofcellshasbeen established.4,6,25,27Preventing TBP-or c-Myc-mediated increases in Pol III gene transcription abrogates their transforming activity.27Greater Pol III gene transcription results in the preferential translation of mRNAs that encode proteins that are required for cell growth,suggesting that the increases in transcription and protein synthesis are not uniform.24The ability of JNK1 to induce Pol III genes supports its function in promoting an oncogenic state,whereas that of JNK2 to impede the expression of this class is consistent with its role in suppressing a transformed phenotype.
3.5.Alcohol-activated JNK1 increases c-Jun activity to augment Pol III gene transcription
Alcohol has been classi fied as a carcinogen in humans.33,34Alcohol-induced liver injury,including liver steatosis,in flammation, fibrosis,and cirrhosis,increases the risk of the development of HCC.87-89Alcohol dehydrogenase(ADH)-mediated metabolism of alcohol is required to stimulate Pol III gene transcription.6Alcohol metabolism by ADH affects endoplasmic reticulum stress.90Activation of the stress kinase JNK1 is necessary for alcohol to induce Pol III gene expression.JNK1 stimulation promotes steatosis and liver damage.40Alcohol dramatically activates JNK1,but not JNK2,in HepG2-ADH cells.6
Alcohol-mediated activation of JNK1 upregulates c-Jun and enhances the activity of AP-1.This pathway increases TBP and Brf1 expression.TBP and Brf1 are limiting components for Pol III gene transcription in hepatocytes,and upmodulation of these proteins is required for alcohol to induce transcription of such genes(Fig.2).6Alcohol-induced TBP and Brf1 expression are co-regulated through greater expression of c-Jun.6Alcohol-mediated induction of AP-1 activity occurs through the enhanced expression of c-Jun but not c-Fos.81,91JNK1 mediates an increase in cellular TBP and Brf1 through the activation and recruitment of Elk-1 to the TBP and Brf1 promoters.8,49Although alcohol stimulates Elk-1 and AP-1,its ability to activate TBP and Brf1 is driven primarily by greater c-Jun expression and its recruitment to their promoters.Downregulation of Elk-1 suppresses the tumorigenicity of HCC cells,50and Elk-1 activation is likely to regulate at least a subset of genes that participate in alcohol-mediated liver disease.Interestingly,we found that Elk-1,c-Jun,and c-Fos occupy sites near the tRNALeugene.6
Genomewide studies indicate that many RNA Pol II transcription factors are associated with Pol III genes and occupy some of the same sites as RNA Pol III.92Importantly,alcohol increases c-Jun occupancy of the tRNA gene,suggesting that it has a direct role in regulating Pol III genes.Thus,it is possible that alcohol-dependent increases in c-Jun upregulate TFIIIB subunits and act directly on Pol III gene promoters to facilitate their transcription.Together,these studies demonstrate that JNKs have pleiotropic effects on gene expression,altering the biosynthetic capacity of cells and thus resulting in changes in cellular phenotypes(Fig.2).
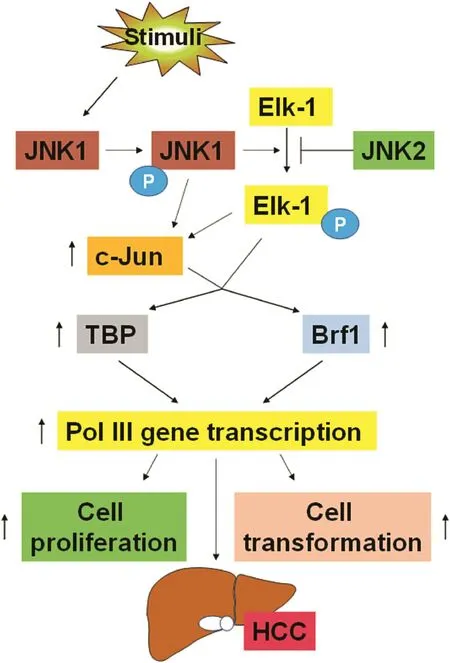
Fig.2.Signaling events of stimulator-induced liver tumor development.Stimulators induce JNK1 activation to upregulate c-Jun expression through Elk-1.Elk-1 and c-Jun augment TBP and Brf1 expression to upregulate Pol III gene transcription,resulting in cell proliferation and cell transformation and eventual HCC development.Blue P:phosphorylation group.Abbreviations:Pol III,RNA polymerase III;TBP,TATA boxbinding protein;Brf1,TFIIB-related factor 1;JNKs,c-Jun amino-terminal kinase;HCC,hepatocellular carcinoma.
4.Epigenetic regulation of Brf1 and Pol III genes by phosphorylated histone 3
4.1.Modi fi ed histone H3 regulates gene activity
Emerging evidence indicates that epigenetic modi fications(eg,phosphorylation,acetylation,methylation,and ubiquitination)of histones are involved in embryonic development,cellular growth,differentiation,proliferation,transformation,and cancer development.However,little is known about whether histone H3 modi fications mediate Pol III gene transcription.Because Pol III gene transcription is linked to cancer development,the mechanism by which H3 modi fications regulate Pol III gene activities must be determined.
The maintenance of a repressed or activated state of a gene is often necessary for embryonic development,cellular differentiation,and pathological conditions and might be governed by a histone code.93In hepatoma cells that were synchronized in a cell cycle time course experiment,histone H3 lysine 4 di-or trimethylation (H3K4me2/me3),histone H3 lysine 79 dimethylation(H3K79me2),and H3 and H4 acetylation were stable at the HNF-1,HNF-4,and albumin promoters.94Global histone modi fication patterns for histone H3 lysine 9 acetylation(H3K9ac),H3K18ac,H4K12ac,H3K4me2,and H4R3me2 indicated that these patterns predicted the risk of prostate cancer recurrence.95
The combination of H3K18ac,H3K9ac,and H3K27ac has been linked togenes that are involved inprotein synthesis.96In humanT-cells,by gene ontology analysis,genes that have been implicated in cell physiology and metabolism are enriched at active promoters and have the 17 backbone modi fications and additional histone modi fications,whereas genes that are involved in cell signaling and other non-T-cell functions,such as development and synaptic transmission,amass at inactive promoters.97,98In addition,H3K9ac,H3K14ac,and H3K4me3 are associated with active genes that are required for T-cell function and development,whereas H3K27me3 correlates with silent genes that participate in the development of other cell types.99
H3K64me3 localizes preferentially to repressive chromatin and may help secure the nucleosome in an appropriately repressed state during development.100In embryonic stem cells,these genes,in addition to tumor suppressor genes and pro-differentiation factors,are held in a “transcription-ready”state by a “bivalent”promoter chromatin pattern thatconsistsofthe repressive H3K27me and active H3K4me markers.101,102Embryonic carcinoma cells also bear two key repressivemarkers,H3K9me2and H3K9me3.Our study indicates that UV-induced histone H3 serine 28 phosphorylation(H3S28ph)increases H3K9ac levels,whereas H3K9ac does not affect H3S28ph,103suggesting that certain H3 modi fications facilitate another type of H3 marker.This effect can be uni-or bidirectional.
4.2.Phosphorylated H3 mediates Brf1 expression and Pol III gene transcription
Histone H3 serine 10 phosphorylation(H3S10ph)and H3S28ph are mediated by MAP kinases,104,105which are involved in mitosis.106,107MSK1,a downstream component of MAP kinase signaling,mediates phosphorylation of histone H3(H3ph).105Sustained JNK1 activation is associated with H3K4me3 and H3K9me3 in human HCC,108and MAP kinase(JNKs,ERKs,p38)stimulation is linked to cancer development.These data indicate that epigenetic modi fications to histones mediate Pol II genes(encoding mRNA).
RNA Pol III transcribes small untranslated tRNAs and 5S rRNA,which participate in protein synthesis.Because these species are smaller and more abundant,Pol III gene transcription re flects their gene activity.It is unknown whether H3 modi fications mediate Pol III gene transcription.Recent studies have indicated that most active chromatin marks in Pol II genes are appear in active Pol III genes,although their exact distribution differs with respect to Pol III genes.109-112But,it implies that epigenetic modi fications of histones regulate Pol III genes.
We have demonstrated that EGF-induced H3S28ph modulates endogenous tRNA and 5S rRNA gene transcription.30EGF-induced H3S28ph occurs through EGFR but not the PI3K pathway.EGF-induced occupancy of the Brf1 and TBP promoters by H3S28ph modulates their expression,which indirectly regulates tRNA and 5S rRNA transcription.30In addition,H3S28ph occupies tRNA and 5S rRNA promoters to directly govern their transcription.30
H3S28ph-mediated Brf1 expression and Pol III gene transcription are critical for cell transformation.Blocking H3S28ph is suf ficient to inhibit tRNA and 5S rRNA transcription and anchorageindependent growth of mouse JB6 cells.30These findings support the novel hypothesis that H3 modi fications,such as H3S28ph,regulate Pol III gene activity.
DEN-induced liver tumor formation is one of the most widely accepted experimentalmodels forstudying hepatocarcinogenesis.113DEN-induced H3S10ph levels in tumor hepatocytes are higher than in normal hepatocytes.28DEN affects H3S10ph and augments Brf1 expression and Pol III gene transcription.28Greater H3S10ph levels in the Brf1 promoter due to DEN modulate its expression to indirectly regulate tRNA and 5S rRNA transcription.28In addition,H3S10ph accumulates in tRNA and 5S rRNA promoters to directly regulate their transcription.Inhibition of H3S10ph is suf ficient to downregulate Brf1 and tRNA and 5S rRNA transcription,impeding cell proliferation and cell transformation.28
These studies present a mechanistic analysis that characterizes how H3S10ph modulates tRNA and 5S rRNA transcription and Brf1 expression,showing that DEN induces H3S10ph,resulting in the upregulation of Pol III genes,phenotypic alterations in hepatocytes,and eventually HCC.28Enhancement of Brf1 expression and Pol III gene transcription correlate with HCC in mice,6,25suggesting that Brf1 is a key molecule in the development of HCC.
DEN induces HCC in rodents.Mice that lack JNK1 are less susceptible to DEN-induced haptocarcinogenesis.40JNK1 positively mediates Pol III gene transcription,whereas JNK2 negatively regulates it.49DEN activates JNK1,which induces H3ph.114JNK1 might be a potential target of HCC therapy.115These studies suggest that DEN potentiates Pol III gene transcription.Our studies support that DEN upregulates Brf1 expression and Pol III gene transcription in cell culture models,28implying that DEN-induced hepatocarcinogenesis is associated with upmodulation of Brf1 and Pol III genes.To further examine the function of Brf1 and the epigenetic regulatory events in liver tumorigenesis,mouse models of DEN-induced HCC will be used to detail such eventsin vivo.
In addition,alcohol activates MSK1(unpublished),which induces H3ph.105Alcohol also induces H3ph(unpublished).Inhibition of MSK1 by its specific inhibitor,SB-747651A,decreases Pol III gene transcription caused byalcohol(unpublished)(Fig.3).Furthermore,human biopsystudies indicate that H3S10ph occurs in tumor foci of breast cancer,compared with nontumor tissues.Brf1 and H3S10ph colocalize in the nucleus(unpublished).These studies demonstrate that H3ph modulates Brf1 expression and Pol III gene transcription.
It is possible that a balance in histone 3 modi ficationsin vivocontrols gene expression in various physiological and pathological states.The balance between acetylation and deacetylation of core histones is regulated by histone acetyltransferases(HATs)and histone deacetylases(HDACs).H3ph is mediated by protein kinases and protein phosphatases.Histone methylation is a result of the coordinated interplay between histone lysine methyltransferases and demethylases at specific gene promoters.Removal of acetyl groups byHDACs is associated with transcriptional repression.116,117Treatment of mammalian cells with HDAC inhibitors,such as TSA and trapoxin,upregulates various genes.118,119c-Myc increases acetylated histone H3 levels in tRNALeuand 5S rRNA promoters and the transcription of these genes.120
TSA alsoactivatesMAPKstoinduceH3S28ph,whereas H3S28ph,in turn,facilitates histone H3 acetylation at lysine 9.103Immediate early genes,such asc-Myc,c-Jun,andc-Fos,induce H3ph,121,122implying that c-Myc induction enhances H3ph to elevate the activity oftRNAand5S rRNAgenes.The function of c-Myc in H3ph merits further study.H3S10ph is critical for full transcriptional activation of virus-like 30S elements.123H3S10ph occupancy of thec-Fospromoter mediates gene activities.124
H3K4me1/2/3 isamarkerofgeneactivation,whereas H3K27me3 re flects gene repression.By ChIP sequencing,tRNAgenes harbor high H3K4me1/2/3 and H3Kac levels and little H3K27me3.125Phosphorylation ofH3K27me3S28 occursin response to stress signaling,mitogenic signaling,and retinoic acid(RA)-induced neuronal differentiation.MSK1/2-mediated phosphorylation of H3K27me3S28 activates a subset of polycomb group target genes and modulates the gene expression program that determines cell fate.126EGF decreases H3K27me3 levels in the tRNALeuand 5S rRNA promoters and elevates H3S28ph levels in them,resulting in upregulation of gene transcription.This finding indicates that various epigenetic modi fications of histone H3 synergize in the regulation of tRNA and 5S rRNA genes.These analyses support that changes in the balance of H3 modi fications constitute a mechanism for regulating tRNA and 5S rRNA transcription.H3S28ph and H3S10ph directly occupy the promoters of Pol II genes,such asc-Fos,Brf1,andTBP,and are directly associated with promoters of Pol III genes,such astRNALeuand5S rRNA,28,30suggesting that bothPol IIandPol IIIgene activities are mediated by common H3 modi fications(Fig.3).
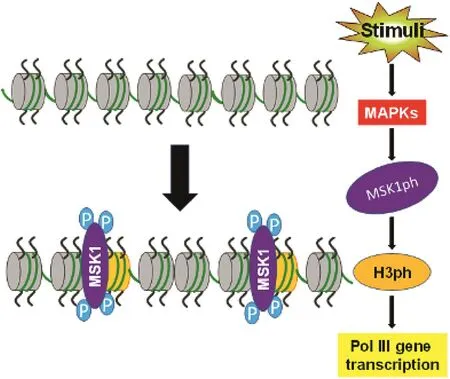
Fig.3.Chromatin remodeling by MSK1-mediated H3ph.These factors activate MAP kinases,which in turn stimulate MSK1 activation by phosphrylated MSK1(MSK1ph).MSK1ph mediates H3ph to affect chromatin remodeling,leading to transcription of Pol III genes.Blue P:phosphorylation group.Abbreviations:MAPKs,mitogen-activated protein kinases;H3ph,histone H3 phosphorylation;Pol III,RNA polymerase III.
5.Conclusions and future perspectives
Upregulation of Brf1 and Pol III genes is tightly linked to cell transformation and tumorigenesis.Our studies have shown that Brf1 expression and Pol III gene transcription are upregulated in human biopsies of HCC and tumor cell lines.JNK1 is a key signaling molecule that is associated with HCC development due to DEN or alcohol,which positively modulates Pol III gene activity through Elk-1 and c-Jun.H3ph induces the expression of these genes.Blocking H3ph decreases Brf1 and Pol III gene transcription and represses cell proliferation and cell transformation.In vivostudies on how H3ph modulates Brf1 and Pol III gene expression need to be performed.The studies that we have presented demonstrate that Brf1 is a key transcription factor that specifically regulates Pol III gene transcription.Thus,inhibiting Brf1 expression might be a potential approach for novel therapies for HCC.
Authors'contributions
S.Zhong conceived of the idea,and J.Lei,S.Chen,and S.Zhong prepared this manuscript.
Con flict of interest
The authors declare that they have no con flict of interest.
Thisworkwassupportedin partbyNIAAA/NIHgrants AA017288,AA021114,AA023247,and AA024169 to SZ.
1.Borchert GM,Lanier W,Davidson BL.RNA polymerase III transcribes human microRNAs.Nat Struct Mol Biol.2006;13:1097-1101.
2.Schramm L,Hernandez N.Recruitment of RNA polymerase III to its target promoters.Genes Dev.2002;16:2593-2620.
3.Shi G,Zhong S.Alcohol-associated cancer and deregulation of Pol III genes.Gene.2017;612:25-28.
4.Zhang Q,Jin J,Zhong Q,Yu X,Levy D,Zhong S.ERαmediates alcohol-induced deregulation of Pol III genes in breast cancer cells.Carcinogenesis.2013;34:28-37.
5.Zhong S,Zheng C,Johnson DL.Epidermal growth factor enhances cellular TBP levels and induces RNA polymerase I-and III-dependent gene activity.Mol Cell Biol.2004;24:5119-5129.
6.Zhong S,Machida K,Tsukamoto H,Johnson DL.Alcohol induces RNA polymerase III-dependent transcription through c-Jun by co-regulating TATA-binding protein(TBP)and Brf1 expression.J Biol Chem.2011;286:2393-2401.
7.Hernandez N.TBP,a universal eukaryotic transcription factor?Genes Dev.1993;7:1291-1308.
8.Zhong S,Johnson DL.The JNKs differentially regulate RNA polymerase III transcription by coordinately modulating the expression of all TFIIIB subunits.Proc Natl Acad Sci U S A.2009;106:12682-12687.
9.White RJ.RNA polymerase III transcription and cancer.Oncogene.2004;23:3208-3216.
10.Johnson DL,Johnson SA.Cell biology,RNA metabolism and oncogenesis.Science.2008;320:461-462.
11.Zhong Q,Shi G,Zhang Y,Lu L,Levy D,Zhong S.Alteration of BRCA1 expression affects alcohol-induced transcription of RNA Pol III-dependent genes.Gene.2015;556:74-79.
12.Woiwode A,Johnson SA,Zhong S,et al.PTEN represses RNA polymerase III-dependent transcription by targeting the TFIIIB complex.Mol Cell Biol.2008;28:4204-4214.
13.Winter AG,Sourvinos G,Allison SJ,et al.RNA polymerase III transcription factor TFIIIC2 is overexpressed in ovarian tumors.Proc Natl Acad Sci U S A.2007;97:12619-12624.
14.Hirsch HA,Jawdekar GW,Lee KA,Gu L,Henry RW.Distinct mechanisms for repression of RNA polymerase III transcription by the retinoblastoma tumor suppressor protein.Mol Cell Biol.2004;24:5989-5999.
15.Sutcliffe JE,Brown TR,Allison SJ,Scott PH,White RJ.Retinoblastoma protein disrupts interactions required for RNA polymerase III transcription.Mol Cell Biol.2000;20:9192-9202.
16.Crighton D,Woiwode A,Zhang C,et al.p53 represses RNA polymerase III transcription by targeting TBP and inhibiting promoter occupancy by TFIIIB.EMBO J.2003;22:2810-2820.
17.Fairley JA,Scott PH,White RJ.TFIIIB is phosphorylated,disrupted and selectively released from tRNA promoters during mitosis in vivo.EMBO J.2003;22:5841-5850.
18.Hu P,Samudre K,Wu S,Sun Y,Hernandez N.CK2 phosphorylation of Bdp1 executes cell cycle-specific RNA polymerase III transcription repression.Mol Cell.2004;16:81-92.
19.Johnson SA,Zheng C,Fromm J,Willis IM,Johnson DL.Mammalian Maf1 is a negative regulator of transcription by all three nuclear RNA polymerases.Mol Cell.2007;26:367-379.
20.Wang HD,Trivedi A,Johnson DL.Hepatitis B virus X protein induces RNA polymerase III-dependent gene transcription and increases cellular TATA-binding protein by activating the Ras signaling pathway.Mol Cell Biol.1997;17:6838-6846.
21.Felton-Edkins ZA,White RJ.Multiple mechanisms contribute to the activation of RNA polymerase III transcription in cells transformed by papovaviruses.J Biol Chem.2002;277:48182-48191.
22.Daly NL,Arvanitis DA,Fairley JA,et al.Deregulation of RNA polymerase III transcription in cervical epithelium in response to high-risk human papillomavirus.Oncogene.2005;24:880-888.
23.Johnson SA,Dubeau L,Kawalek M,et al.Increased expression of TATA-binding protein,the central transcription factor,can contribute to oncogenesis.Mol Cell Biol.2003;23:3043-3051.
24.Marshall L,Kenneth NS,White RJ.Elevated tRNA(iMet)synthesis can drive cell proliferation and oncogenic transformation.Cell.2008;133:78-89.
25.Zhong Q,Xi S,Liang J,et al.The signi ficance of Brf1 overexpression in human hepatocellular carcinoma.Oncotarget.2016;7:6243-6254.
26.Cabarcas S,Jacob J,Vera I,Schramm L.Differential expression of the TFIIIB subunits Brf1 and Brf2 in cancer cells.BMC Mol Biol.2008;9:74.
27.Johnson SA,Dubeau L,Johnson DL.Enhanced RNA polymerase III-dependent transcription is required for oncogenic transformation.J Biol Chem.2008;283:19184-19191.
28.Zhong Q,Shi G,Zhang Q,Zhang Y,Levy D,Zhong S.Role of phosphorylated histone H3 serine 10 in DEN-induced deregulation of Pol III genes and cell proliferation and transformation.Carcinogenesis.2013;34:2460-2469.
29.Yi Y,Huang C,Zhang Y,et al.Exploring a common mechanism of alcoholinduced deregulation of RNA Pol III genes in liver and breast cells.Gene.2017;626:309-318.
30.Zhang Q,Zhong Q,Evans AG,Levy D,Zhong S.Phosphorylation of histone H3 serine 28 modulates RNA polymerase III-dependent transcription.Oncogene.2011;30:3943-3952.
31.Zhong Q,Shi G,Zhang Q,Lu L,Levy D,Zhong S.Tamoxifen represses alcoholinduced transcription of RNA polymerase III-dependent genes in breast cancer cells.Oncotarget.2014;5:12410-12417.
32.Machida K,Tsukamoto H,Mkrtchyan H,et al.Toll-like receptor 4 mediates synergism between alcohol and HCV in hepatic oncogenesis involving stem cell marker Nanog.Proc Natl Acad Sci U S A.2009;106:1548-1553.
33.International Agency for Research on Cancer.IARC monographs on the evaluation of carcinogenic risks to humans.In:A Review of Human Carcinogens.vol.100.Lyon,France:International Agency for Research on Cancer;2011.https://monographs.iarc.fr/ENG/Monographs/vol44/mono44.pdf.
34.Cogliani VJ,Baan R,Straif K,et al.Preventable exposures associated with human cancers.J Natl Cancer Inst.2011;103:1827-1839.
35.Wang S,Xu M,Li F,et al.Ethanol promotes mammary tumor growth and angiogenesis:the involvement of chemoattractant factor MCP-1.Breast Cancer Res Treat.2012;133:1037-1048.
36.Wong AW,Dunlap SM,Holcomb VB,Nunez NP.Alcohol promotes mammary tumor development via the estrogen pathway in estrogen receptor alphanegative HER2/neu Mice.Alcohol Clin Exp Res.2012;36:577-587.
37.Gao J,Aksoy BA,Dogrusoz U,et al.Integrative analysis of complex cancer genomics and clinical pro files using the cBioPortal.Sci Signal.2013;6:pl1.
38.Naugler WE,Sakurai T,Kim S,et al.Gender display in liver cancer due to sex differences in MyD88-dependent IL-6-production.Science.2007;317:121-124.
39.Watanabe T,Tanaka G,Hamada S,et al.Dose-dependent alterations in gene expression in mouseliverinduced bydiethylnitrosamineand ethylnitrosourea and determined by quantitative real-time PCR.Mutat Res.2009;673:9-20.
40.Sakurai T,Maeda S,Chang L,Karin M.Loss of hepatic NF-kappa B activity enhances chemical hepatocarcinogenesis through sustained c-Jun N-terminal kinase 1 activation.Proc Natl Acad Sci U S A.2006;103:10544-10551.
41.Maeda S,Kamata H,Luo JL,Leffert H,Karin M.IKKbeta couple hepatocyte death to cytokine-driven compensatory proliferation that promotes chemical hepatocarcinogenesis.Cell.2005;121:977-990.
42.Chang L,Karin M.Mammalian MAP kinase signaling cascades.Nature.2001;410:37-40.
43.Davis RJ.Signal transduction by the JNK group of MAP kinases.Cell.2000;103:239-252.
44.Derijard B,Hibi M,Wu IH,et al.JNK1:a protein kinase stimulated by UV light and Ha-Ras that binds and phosphorylates the c-Jun activation domain.Cell.1994;76:1025-1037.
45.Kallunki T,Su B,Tsigelny I,et al.JNK2 contains a specificity-determining region responsible for ef ficient c-Jun binding and phosphorylation.Genes Dev.1994;8:2996-3007.
46.Mohit AA,Martin JH,Miller CA.p493F12 kinase:a novel MAP kinase expressed in a subset of neurons in the human nervous system.Neuron.1995;14:67-78.
47.Engelberg D.Stress-activated protein kinases-tumor suppressors or tumor initiators?Semin Cancer Biol.2004;14:271-282.
48.Kennedy NJ,Davis RJ.Role of JNK in tumor development.Cell Cycle.2003;2:199-201.
49.Zhong S,Fromm J,Johnson DL.TBP is differentially regulated by c-Jun N-terminal kinase 1(JNK1)and JNK2 through Elk-1,controlling c-Jun expression and cell proliferation.Mol Cell Biol.2007;27:54-64.
50.Ying TH,Hsieh YH,Hsieh YS,Liu JY.Antisense oligonucleotide Elk-1 suppresses the tumorigenicity of human hepatocellular carcinoma cells.Cell Biol Int.2008;32:210-216.
51.Chang L,Jones Y,Ellisman MH,Goldstein LS,Karin M.JNK1 is required for maintenance of neuronal microtubules and controls phosphorylation of microtubule-associated proteins.Dev Cell.2003;4:521-533.
52.Gupta S,Barrett T,Whitmarsh AJ,et al.Selective interaction of JNK protein kinase isoforms with transcription factors.EMBO J.1996;15:2760-2770.
53.Liu J,Minemoto Y,Lin A.c-Jun N-terminal protein kinase 1(JNK1),but not JNK2,is essential for tumor necrosis factor alpha-induced c-Jun kinase activation and apoptosis.Mol Cell Biol.2004;24:10844-10856.
54.Sabapathy K,Hochedlinger K,Nam SY,Bauer A,Karin M,Wagner EF.Distinct roles for JNK1 and JNK2 in regulating JNK activity and c-Jun-dependent cell proliferation.Mol Cell.2004;15:713-725.
55.Engelberg D.Stress-activated protein kinases-tumor suppressors or tumor initiators?Semin Cancer Biol.2004;14:271-282.
56.Kennedy NJ,Davis RJ.Role of JNK in tumor development.Cell Cycle.2003;2:199-201.
57.Shaulian E,Karin M.AP-1 as a regulator of cell life and death.Nat Cell Biol.2002;4:E131-E136.
58.Fuchs SY,Adler V,Buschmann T,et al.JNK targets p53 ubiquitination and degradation in non-stressed cells.Genes Dev.1998;12:2658-2663.
59.Trivedi A,Vilalta A,Gopalan S,Johnson DL.TATA-binding protein is limiting for both TATA-containing and TATA-lacking RNA polymerase III promoters inDrosophilacells.Mol Cell Biol.1996;16:6909-6916.
60.Wang HD,Trivedi A,Johnson DL.Regulation of RNA polymerase I-dependent promoters by the hepatitis B virus X protein,activated Ras and TATA-binding protein.Mol Cell Biol.1998;18:7086-7094.
61.Wang HD,Yuh CH,Dang CV,Johnson DL.The hepatitis B virus X protein increases the cellular level of TATA-binding protein,which mediates transactivation of RNA polymerase III genes.Mol Cell Biol.1995;15:6720-6728.
62.Colgan J,Manley JL.TFIID can be rate limiting in vivo for TATA-containing but not TATA-lacking,RNA pol II promoters.Genes Dev.1992;6:304-315.
63.Majello B,Napolitano G,De Luca P,Lania L.Recruitment of human TBP selectively activates RNA polymerase II TATA-dependent promoters.J Biol Chem.1998;273:16509-16516.
64.Sadovsky Y,Webb P,Lopez G,et al.Transcriptional activators differ in their responses to overexpression of TATA-box-binding protein.Mol Cell Biol.1995;15:1554-1563.
65.Tsuiki H,Tnani M,Okamoto I,et al.Constitutively active forms of c-Jun NH2-terminal kinase are expressed in primary glial tumors.Cancer Res.2003;63:250-255.
66.Johnson SA,Dubeau L,White RL,Johnson DL.The TATA-binding protein as a regulator of cellular transformation.Cell Cycle.2003;2:442-444.
67.Cavigelli M,Dol fiF,Claret FX,Karin M.Induction of c-fos expression through JNK-mediated TCF/Elk-1 phosphorylation.EMBO J.1995;14:5957-5964.
68.Whitmarsh AJ,Shore P,Sharrocks AD,Davis RJ.Integration of MAP kinase signal transduction pathways at the serum response element.Science.1995;269:403-407.
69.Yang SH,Shore P,Willingham N,Lakey JH,Sharrocks AD.The mechanism of phosphorylation-inducible activation of the ETS-domain transcription factor Elk-1.EMBO J.1999;18:5666-5674.
70.Li QJ,Yang SH,Maeda Y,Sladek FM,Sharrocks AD,Martins-Green M.MAP kinase phosphorylation-dependent activation of Elk-1 leads to activation of the co-activator p300.EMBO J.2003;22:281-291.
71.Goodfellow SJ,Innes F,Derblay LE,MacLellan WR,Scott PH,White RJ.Regulation of RNA polymerase III transcription during hypertrophic growth.EMBO J.2006;25:1522-1533.
72.Yang SH,Whitmarsh AJ,Davis RJ,Sharrocks AD.Differential targeting of MAP kinases to the ETS-domain transcription factor Elk-1.EMBO J.1998;17:1740-1749.
73.Treisman R.Regulation of transcription by MAP kinase cascades.Curr Opin Cell Biol.1996;8:205-215.
74.Tian J,Karin M.Stimulation of Elk1 transcriptional activity by mitogenactivated protein kinases is negatively regulated by protein phosphatase 2B(calcineurin).J Biol Chem.1999;274:15173-15180.
75.Jaeschke A,Karasarides M,Ventura JJ,et al.JNK2 is a positive regulator of the c-Jun transcription factor.Mol Cell.2006;23:899-911.
76.Tournier C,Hess P,Yang DD,et al.Requirement of JNK for stress-induced activation of the cytochrome c-mediated death pathway.Science.2000;288:870-874.
77.Dunn C,Wiltshire C,MacLaren A,Gillespie DA.Molecular mechanism and biological functions of c-Jun N-terminal kinase signaling via the c-Jun transcription factor.Cell Signal.2002;14:585-593.
78.Fuchs SY,Dolan L,Davis RJ,Ronai Z.Phosphorylation dependent targeting of c-Jun ubiquitination by Jun N-kinase.Oncogene.1996;13:1531-1535.
79.Choi BY,Choi HS,Ko K,et al.The tumor suppressor p16(INK4a)prevents cell transformation through inhibition of c-Jun phosphorylation and AP-1 activity.Nat Struct Mol Biol.2005;12:699-707.
80.Tafolla E,Wang S,Wong B,Leong J,Kapila YL.JNK1 and JNK2 oppositely regulate p53 in signaling linked to apoptosis triggered by an altered fibronectin matrix:JNK links FAK and p53.J Biol Chem.2005;280:19992-19999.
81.Zhong Q,Shi G,Zhang Y,Levy D,Zhong S.Elk-1 and AP-1 sites in the TBP promoter mediate alcohol-induced deregulation of Pol III-dependent genes.Gene.2013;526:54-60.
82.Hess P,Pihan G,Sawyers CL,Flavell RA,Davis RJ.Survival signaling mediated by c-Jun NH(2)-terminal kinase in transformed B lymphoblasts.Nat Genet.2002;32:201-205.
83.She QB,Chen N,Bode AM,Flavell RA,Dong Z.De ficiency of c-Jun-NH(2)-terminal kinase-1 in mice enhances skin tumor development by 12-O-tetradecanoylphorbol-13-acetate.Cancer Res.2002;62:1343-1348.
84.Chen N,Normura M,She QB,et al.Suppression of skin tumorigenesis in c-Jun NH(2)-terminal kinase-2-de ficient mice.Cancer Res.2001;61:3908-3912.
85.Mayer C,Bierhoff H,Grummt I.The nucleolus as a stress sensor:JNK2 inactivates the transcription factor TIF-IA and down-regulates rRNA synthesis.Genes Dev.2005;19:933-941.
86.Marshall L,White RJ.Non-coding RNA production by RNA polymerase III is implicated in cancer.Nat Rev Cancer.2008;8:911-914.
87.Lieber CS.Hepatic,metabolic,and nutritional disorders of alcoholism:from pathogenesis to therapy.Crit Rev Clin Lab Sci.2000;37:551-584.
88.Seitz HK,Po¨schi G,Simanowski UA.Alcohol and cancer.Recent Dev Alcohol.1998;14:67-95.
89.Seitz HK,Stickel F.Molecular mechanisms of alcohol-mediated carcinogenesis.Nat Rev Cancer.2007;7:599-612.
90.Nishitani Y,Matsumoto H.Ethanol rapidly causes activation of JNK associated with ER stress under inhibition of ADH.FEBS Lett.2006;580:9-14.
91.Yeligar SM,Machida K,Tsukamoto H,Kalra VK.Ethanol augments RANTES/CCL5 expression in rat liver sinusoidal endothelial cells and human endothelial cells via activation of NF-kappa B,HIF-1 alpha,and AP-1.J Immunol.2009;183:5964-5976.
92.Raha D,Wang Z,Moqtaderi Z,et al.Close association of RNA polymerase II and many transcription factors with Pol III genes.Proc Natl Acad Sci U S A.2010;107:3639-3644.
93.Felsenfeld G,Groudine M.Controlling the double helix.Nature.2003;421:448-453.
94.Kouskouti A,Talianidis I.Histone modi fications de fining active genes persist after transcriptional and mitotic inactivation.EMBO J.2005;24:347-357.
95.Seligson DB,Horvath S,Shi T,et al.Global histone modi fication patterns predict risk of prostate cancer recurrence.Nature.2005;435:1262-1266.
96.Kurdistani SK,Tavazoie S,Grunstein M.Mapping global histone acetylation patterns to gene expression.Cell.2004;117:721-733.
97.Wang Z,Zang C,Rosenfeld JA,et al.Combinatorial patterns of histone acetylations and methylations in the human genome.Nat Genet.2008;40:897-903.
98.Lennatsson A,Ekwall K.Histone modi fication patterns and epigenetic codes.Biochim Biophys Acta.2009;1790:863-868.
99.Ruh TY,Cuddapah S,Cui K,Zhao K.The genomic landscape of histone modi fications in human T cells.Proc Natl Acad Sci U S A.2006;103:15782-15787.100.Daujat S,Weiss T,Mohn F,et al.H3K64 trimethylation marks heterochromatin and is dynamically remodeled during developmental reprogramming.Nat Struct Mol Biol.2009;16:777-781.
101.Ohm JE,McGarvey KM,Yu X,et al.A stem cell-like chromatin pattern may predispose tumor suppressor genes to DNA hypermethylation and heritable silencing.Nat Genet.2007;39:237-242.
102.Eissenberg JC,Shilatifard A.Histone H3 lysine 4(H3K4)methylation in development and differentiation.Dev Biol.2010;339:240-249.
103.Zhong S,Goto H,Inagaki M,Dong Z.Phosphorylation at serine 28 and acetylation at lysine 9 of histone H3 induced by trichostatin A.Oncogene.2003;22:5291-5297.
104.Zhong SP,Ma WY,Dong Z.ERKs and p38 kinases mediate ultraviolet B-induced phosphorylation of histone H3 at serine 10.J Biol Chem.2000;275:20980-20984.
105.Zhong S,Jansen C,She QB,et al.Ultraviolet B-induced phosphorylation of histone H3 at serine 28 is mediated by MSK1.J Biol Chem.2001;276:33213-33219.
106.Goto H,Tomono Y,Ajiro K,et al.Identi fication of a novel phosphorylation site on histone H3 coupled with mitotic chromosome condensation.J Biol Chem.1999;274:25543-25549.
107.Wei Y,Yu L,Bowen J,Gorovsk MA,Allis CD.Phosphorylation of histone H3 is required forproperchromosomecondensation and segregation.Cell.1999;97:99-109.
108.Chang Q,Zhang Y,Beezhold KJ,et al.Sustained JNK1 activation is associated with altered histone H3 methylations in human liver cancer.J Hepatol.2009;50:323-333.
109.Raha D,Wang Z,Mogtaderi Z,et al.Close association of RNA polymerase II and many transcription factors with Pol III genes.Proc Natl Acad Sci U S A.2010;107:3639-3644.
110.Barski A,Chepelev I,Liko D,et al.Pol II and its associated epigenetic marks are present at Pol III-transcribed noncoding RNA genes.Nat Struct Mol Biol.2010;17:629-634.
111.Oler AJ,Alla RK,Roberts DN,et al.Human RNA polymerase III transcriptomes and relationships to Pol II promoter chromatin and enhancer-binding factors.Nat Struct Mol Biol.2010;17:620-628.
112.Moqtaderi Z,Wang J,Raha D,et al.Genomic binding profiles of functionally distinct RNA polymerase III transcription complexes in human cells.Nat Struct Mol Biol.2010;17:635-640.
113.Ha WS,Kim CK,Song SH,Kang CB.Study on mechanism of multistep hepatotumorigenesis in rat:development of hepatotumorigenesis.J Vet Sci.2001;2:53-58.
114.Zhong S,Zhang Y,Jansen C,Goto H,Inagaki M,Dong Z.MAP kinases mediated UVB-induced phosphorylation of histone H3 at serine 28.J Biol Chem.2001;276:12932-12937.
115.Chen F,Beezhold K,Castranova V.JNK1,a potential therapeutic target for hepatocellular carcinoma.Biochim Biophys Acta.2009;1796:242-251.
116.Struhl K,Moqtaderi Z.The TAFs in the HAT.Cell.1998;94:1-4.
117.Finnin MS,Donigian JR,Cohen A,et al.Structures of a histone deacetylase homologue bound to the TSA and SAHA inhibitors.Nature.1999;401:188-193.
118.Kijima M,Yoshida M,Sugita K,Horinouchi S,Beppu T.Trapoxin,an antitumor cyclic tetrapeptide,is an irreversible inhibitor of mammalian histone deacetylase.J Biol Chem.1993;268:22429-22435.
119.Yoshida M,Horinouchi S.Trichostatin and leptomycin.Inhibition of histone deacetylation and signal-dependent nuclear export.Ann N Y Acad Sci.1999;886:23-36.
120.Kenneth NS,Ramsbottom BA,Gomez-Roman N,Marshall L,Cole PA,White RJ.TRRAP and GCN5 are used by c-Myc to activate RNA polymerase III transcription.Proc Natl Acad Sci U S A.2007;104:14917-14922.
121.Cheung P,Tanner KG,Cheung WL,Sassone-Corsi P,Denu JM,Allis CD.Synergistic coupling of histone H3 phosphorylation and acetylation in response to epidermal growth factor stimulation.Mol Cell.2000;5:905-915.
122.Wei Y,Yu L,Bowen J,Gorovsky MA,Allis CD.Phosphorylation of histone H3 is required forproperchromosomecondensation and segregation.Cell.1999;97:99-109.
123.Brunmeir R,Lagger S,Simboeck E,et al.Epigenetic regulation of a murine retrotransposon by a dual histone modi fication mark.PLoS Genet.2010;6:e1000927.
124.Shimada M,Nakadai T,Fukuda A,Hisatake K.cAMP-response elementbinding protein(CREB)controls MSK1-mediated phosphorylation of histone H3 at the c-fos promoter in vitro.J Biol Chem.2010;285:9390-9401.
125.Barki A,Chepelev I,Liko D,et al.Pol II and its associated epigenetic marks are present at Pol III-transcribed noncoding RNA genes.Nat Struct Mol Biol.2010;17:629-634.
126.Gehani SS,Agrawal-Singh S,Dietrich N,Christophersen NS,Helin K,Hansen K.Polycomb group protein displacement and gene activation through MSK-dependent H3K27me3S28 phosphorylation.Mol Cell.2010;39:886-900.
杂志排行

Liver Research的其它文章
- Mesenchymal stem cells for treatment of steroid-resistant acute rejection after liver transplantation☆
- Stopping nucleos(t)ide analog treatment in chronic hepatitis B-Who and when?☆
- Wilson disease:At the crossroads between genetics and epigenetics-A review of the evidence☆
- Methionine adenosyltransferases in liver health and diseases☆
- Activating transcription factor 3 in immune response and metabolic regulation☆
- Human embryoid bodies to hepatocyte-like clusters:Preparing for translation☆