带电粒子对FOX-7裂解通道影响的密度泛函研究
2017-05-07李小东王晶禹王新全
李小东, 徐 哲, 王晶禹, 王新全
(中北大学化工与环境学院, 山西 太原 030051)

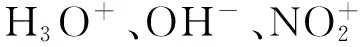
2 计算方法
采用密度泛函理论的B3LYP、B3PW91和PBE0(即Gaussian 03中的PBE1PBE)三种泛函理论,在6-31++G(d,p)基组下,对各反应通道上的反应物、中间体、过渡态和产物进行全参数优化,得到最优几何构型; 并采用自然键轨道(NBO)理论分析了相关结构的键级和成键方式。在相同的水平下对所有结构进行频率分析,使反应物、中间体和产物在势能面的极小值点,过渡态在一阶鞍点。所有活化能计算均经过零点能(ZPE)校正,所有过渡态采用内禀反应坐标(IRC)计算,以确认每个过渡态连接的2个极小点是相应的反应物和产物。所有电子结构及能量计算均采用Gaussian 03程序包[9]。
3 结果与讨论
3.1 构型优化与静电势分析
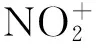
图1 在B3LYP/6-31++G(d,p)水平下优化的各带电粒子与FOX-7复合体的构型
Fig.1 Optimized configurations of the H3O+, OH-and FOX-7 complexes at B3LYP/6-31++G(d,p) level
由图1可知,H3O+和OH-通过直接成键的方式与FOX-7结合。H3O+与FOX-7接近后,H+脱离H2O后连接在FOX-7的不同位置,使分子内的电子结构重新排布,形成了H3O+_01和H3O+_02。OH-与FOX-7接近后,若夺取氨基上的一个质子,则FOX-7变成了带负电的OH-_02构型,若OH-直接进攻C(1)(连接氨基的为C(1),连接硝基的为C(2),下同),则形成了OH-_01。
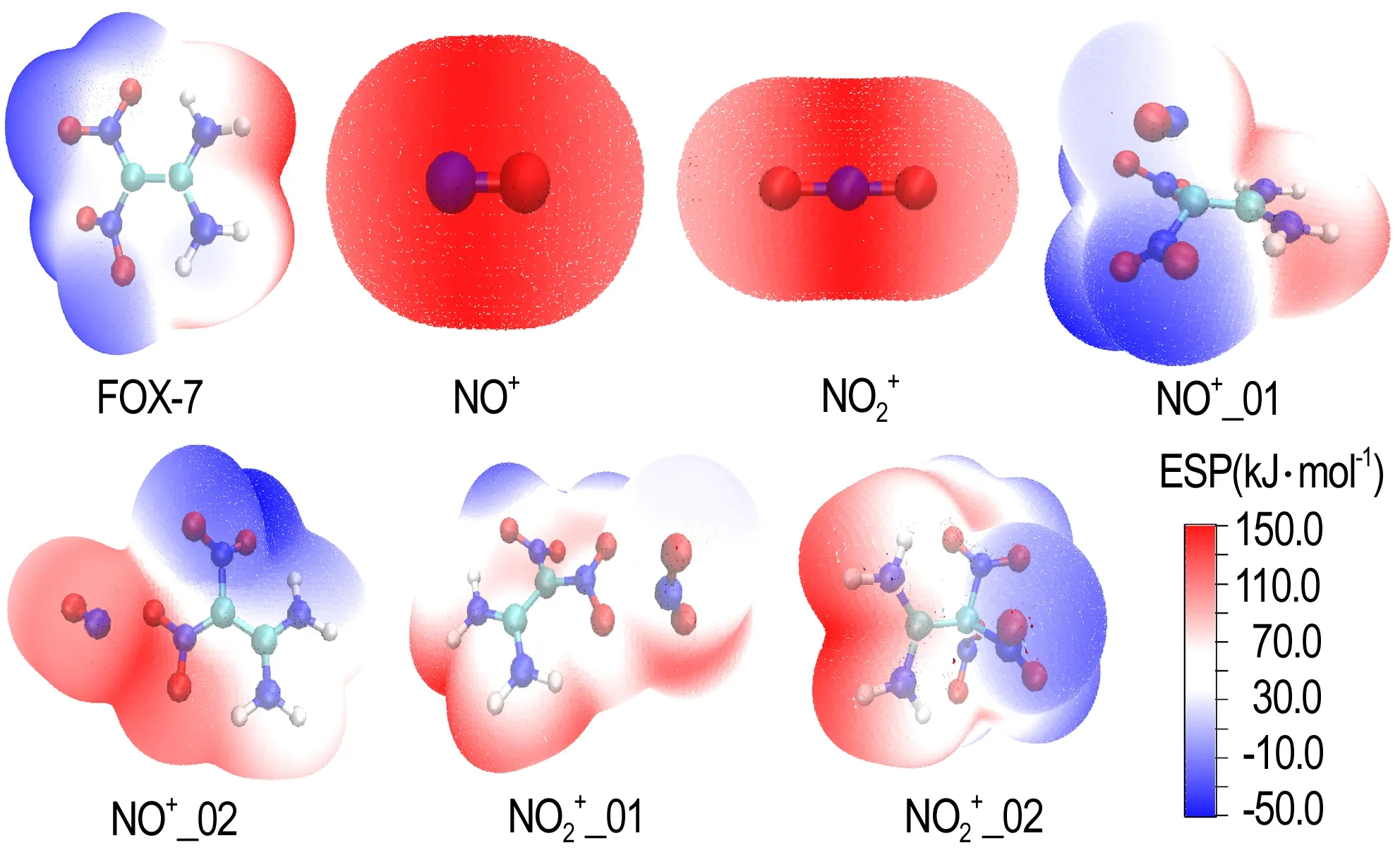


不论是直接成键,还是弱相互作用导致静电势改变,其本质都是由于带电粒子与FOX-7接触后,原有电子排布受到扰乱,从而改变了FOX-7原有的电子结构,而电子结构的改变必然会直接影响键的裂解能等反应性能。
3.2 活化能及势能面
使用密度泛函理论计算在四种带电粒子的影响下各反应通道活化能的变化,并构建各反应通道的势能面。具有明显过渡态的反应,活化能为过渡态与反应物的能量差。当反应为生成自由基的键断裂反应时求其离解能(BDE),此时离解能亦可视为特殊的活化能[13],首先对键断裂后形成的两部分分别进行全参数优化,经频率分析零点能校正后,得到两部分的能量之和,再与键断裂之前的能量做差可得到离解能。
表1为采用B3LYP、B3PW91和PBE0三种泛函,在6-31++G(d,p)基组下计算所得各构型不同裂解通道的活化能,可以看到三种泛函所得数据基本一致,对计算结论无实质性影响,以下讨论未经说明时均为B3LYP/6-31++G(d,p)水平。从表1可知C—NO2键裂解和硝基异构的活化能与文献报道[6]基本一致,证明计算模型与计算方法可行。
表1 不同泛函计算所得活化能
Table 1 Activation energy obtained by different functionals

kJ·mol-1
图3是FOX-7与H3O+复合构型裂解通道的势能面,图3中将H3O+_01、H3O+_02构型的能量设定为0,绿线表示两种构型裂解的最低能量路径,C—C_01表示H3O+_01的C—C键裂解,isomer_02表示H3O+_02的硝基异构,下同。
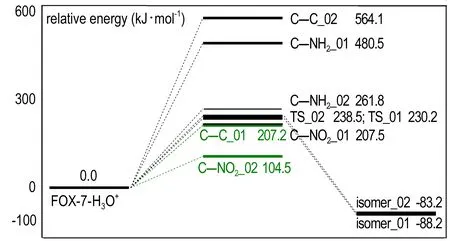
图3 FOX-7与H3O+复合体裂解通道的势能剖面
Fig.3 Potential energy surface of FOX-7-H3O+complex dissociation channels
由图3可知,与单分子FOX-7裂解相比,H3O+_01中活化能下降最多的通道为C—C键离解,经大幅度降低271.5 kJ·mol-1后,C—C键离解成为H3O+_01裂解的最小能量路径。在H3O+_01中,C—C键由FOX-7中的1.426 Å增长到1.459 Å,而周围的2个C—NH2键均减小了0.01 Å,与H3O+作用的硝基所在的C—N键更是缩小了0.08 Å。NBO分析可知,在FOX-7单分子中C—C键成键方式为C1 sp1.7+C2 sp1.44和C1 p+C2 p,C—C键之间总共束缚了3.654个电子。而在H3O+_01中C—C键成键方式为C1 sp1.89+C2 sp1.45,总共束缚了1.97个电子。H3O+的存在使原本被束缚在C—C键中的电子被正电荷吸引而转移。正是由于这些因素综合作用才使原本很难裂解的C—C键成为H3O+_01中最易裂解的键。
H3O+_02构型是H+脱离H2O后进攻C(2)形成的,其中C—C键增长了0.096 Å,两个C—NO2键分别增长了0.122 Å和0.088 Å,而C—NH2键则缩短了0.031 Å。在单分子FOX-7中,C—C键的Wiberg键级为1.217,C—NO2键的键级为1.07,C—NH2键的键级为1.477。而在H3O+_02中,三个键级分别变为0.97,0.91,和1.65。可以看出,C—NO2键的键长增长最多,键级最小。从而,C—NO2键的均裂成了H3O+_02构型裂解的最小能量通道,裂解能为104.5 kJ·mol-1,比单分子FOX-7降低了157.1 kJ·mol-1。
图4为FOX-7与OH-复合体裂解通道的势能剖面,由图4可知,在OH-_01的裂解中只有C—NH2键离解能是下降的,其余三种反应通道的能垒都有不同程度的提高。在单分子FOX-7中C—NH2键的组成为sp2.18+sp1.8,而在OH-_01中为sp2.88+sp2.36。显然在OH-_01的C—NH2键中,p轨道成分增多,s轨道成分减少,轨道由sp2杂化向sp3过渡,电子离域性较大的π键消失,变为单纯的σ键。因此,在OH-_01中C—NH2键的离解能降低。
OH-夺取FOX-7氨基上的一个质子,剩余的就是整体带负电的OH-_02。此结构相当于FOX-7失去了一个H+,多余的一个负电荷被本来位于正电势区域的氨基吸引而重新分布,形成了离域电子。化学键的本质就是电子被束缚在原子核之间,并与之相互作用,所以离域电子的存在使分子结构更加稳定。除硝基异构的活化能稍有减小外,其余三类反应的能垒均有大幅提高。其中C—C键均裂能垒甚至提高了193.4 kJ·mol-1。

图4 FOX-7与OH-复合体裂解通道的势能剖面
Fig.4 Potential energy surface of FOX-7-OH-complex dissociation channels
在NO+_01中NO+的氮原子与C(2)的距离为2.086 Å,NO+是通过弱相互作用与FOX-7复合的。在NO+的影响下,C—NO2键的键长从1.433 Å增长到1.469 Å,原先集中于C—NO2基团的电子在NO+的吸引下向C(2)偏移,使硝基几乎脱离了原FOX-7结构,C—NO2键裂解能仅为4.8 kJ·mol-1,如图5所示。NO+对原本就位于正电势区域的C—NH2键影响不大,仅使裂解能下降了5 kJ·mol-1。电子向C(2)转移使C—C键增强,裂解能提高了77.6 kJ·mol-1。在NO+_02中,NO+中的氮原子接近硝基上的氧原子,距离为1.878 Å,同样为弱相互作用。在NO+的影响下使FOX-7分子中的负静电势区域向一侧的硝基集中,受其影响,临近的氨基也呈现部分的负静电势性质,这使氨基的C—N键具有了部分离域π键的性质,离解能提高。
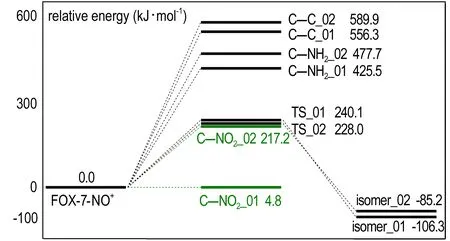
图5 FOX-7与NO+复合体裂解通道的势能面
Fig.5 Potential energy surface of FOX-7-NO+complex dissociation channels
由上述分析可知,虽然四种带电粒子对各裂解通道的活化能影响不尽相同,但可以看出,在所有构型中硝基异构通道的活化能变化都不大,大多约20 kJ·mol-1。金朋刚等[14]在采用原位热红外光谱技术对 FOX-7 热解过程的产物进行原位在线检测时发现,反应初期NO最先出现,随后NO2等产物才出现,但之后NO2的含量急剧增长,很快就超过了NO。这表明FOX-7裂解的引发反应为硝基异构,裂解中期C—NO2键均裂成为主反应通道。宗和厚等[15]计算FOX-7不同裂解通道的速率常数时也发现,随着反应的进行C—NO2键均裂的速率常数很快超过了硝基异构的速率常数。从本文计算结果来看,反应初期硝基异构为最小能量通道,首先反应释放NO; 反应中期随着各种裂解产物的出现,C—NO2的离解能大幅减小,而硝基异构的活化能受后续反应产物的影响较小,所以裂解中期C—NO2键均裂成为主反应通道,理论计算与实验结果[14]一致。


3.3 硝基异构过渡态分析
根据过渡态理论,反应过渡态位于势能面的一阶鞍点,一端连着反应物,另一端连着产物,处于化学键断裂和生产的中间状态,能量比反应物和产物都高。由此可知过渡态在反应中扮演着重要的角色,分析过渡态的结构可深入认识反应的过程,若过渡态结构与反应和产物均很接近,则活化能必然较低; 否则,活化能就高。由上述计算可知,硝基异构通道的活化能受带电粒子的影响较小,采用B3LYP泛函时活化能为220~250 kJ·mol-1,与单分子FOX-7的裂解时硝基异构的活化能(246.1 kJ·mol-1)没有明显差异,硝基异构通道活化能相对稳定的性质与其对应的过渡态结构必然相关。在所计算的四种裂解通道中,硝基异构是唯一具有明显过渡态的裂解通道,图7是在B3LYP/6-31++G(d,p)水平下所得各复合体硝基异构通道的过渡态结构。由图7可知,8种复合体硝基异构通道的过渡态结构及其相似,C—N键的键长集中在1.50~1.58 Å,C—O键的键长在1.71~1.86 Å,硝基中的O—N与FOX-7的C—C轴的夹角基本都为90°,过渡态结构的相似就是活化能基本一致的本质原因。
从过渡态结构还能推测异构反应的动态过程,以H3O+_01硝基异构为例,在H3O+_01中,发生异构的C—NO2键的键长为1.48 Å,在C—NO2片段中C原子与O原子的距离为2.31 Å,硝基中的O—N与FOX-7的C—C轴夹角为44.5°。而在H3O+_01硝基异构的过渡态中,C—NO2键的键长为1.58 Å,C原子与O原子的距离为1.74 Å, O—N与C—C轴夹角为87.6°。由此可知硝基异构的过程为,首先硝基片段以C—N键为轴旋转43.1°,然后C—N键从1.48 Å伸长到1.58 Å的同时C原子与O原子的距离从2.31 Å缩短到1.74 Å,形成过渡态,之后C—N键彻底断裂,C—N键形成,产物逐渐生产。

图7 在B3LYP/6-31++G(d,p)水平下所得各复合体硝基异构通道的过渡态结构
Fig.7 The transition state structure of the nitro isomerism channel of each complex at B3LYP/6-31++G(d,p) level
4 结 论
(3)在所计算的四种裂解通道中硝基异构的活化能受带电粒子的影响最小,在采用B3LYP泛函时活化能在220~250 kJ·mol-1之间,与单分子FOX-7的裂解时硝基异构的活化能(246.1 kJ·mol-1)差异不大,由过渡态分析可知,这是由各复合体硝基异构通道的过渡态具有极其相似的几何构型所致。
综上所述,带电粒子的存在扰乱了原本相对稳定的FOX-7分子共轭体系,使部分基团的键被加强,而部分键被弱化。而对于炸药分子裂解来说,只要有一个键被弱化就相当于催化了裂解反应。从一个全新的视角审视FOX-7裂解中后期的自加速过程,更进一步可以尝试解释炸药分子“相对稳定”的化学本质。
参考文献:
[1] Nikolai V L, Jan B, Abraham L, et al. Synthesis and reaction of 1,1-diamino-2,2-dinitroethylene[J].Tetrahedron, 1998, 54: 11525-11536.
[2] 付秋菠, 舒远杰, 黄奕刚, 等. FOX-7晶体的制备和热性质[J]. 火炸药学报, 2009, 32(04): 6-9.
FU Qiu-bo, SHU Yuan-jie, HUANG Yi-gang, et al. Preparation and thermal properties of FOX-7[J].ChineseJournalofExplosives&Propellants, 2009, 32(04): 6-9.
[3] 付小龙, 樊学忠, 李吉祯, 等. FOX-7研究新进展[J]. 科学技术与工程, 2014, 14(14): 112-119.
FU Xiao-long, FAN Xue-zhong, LI Ji-zhen, et al. Thenew progress of FOX- 7[J].ScienceTechnologyandEngineering, 2014, 14(14): 112-119.
[4] Peter P, Monica C C, Grice M E, et al. Computational investigation of the structures and relative stabilities of amino/nitro derivatives of ethylene[J].JournalofMolecularStructure(TheoChem), 1998, 452: 75-83.
[5] Helen D. Computational studies of FOX-7, a new insensitive explosive, DSTO-TR-1054[R], 2000.
[6] Asta G, Lou M, Lu L H, et al. Proposed mechanism of 1,1-diamino-dinitroethylene decomposition: a density functional theory study[J].TheJournalofPhysicalChemistryA, 1999, 103: 11045-11051
[7] Martin C, Svatopluk C, Kristyna S, et al. Laser ablation of FOX-7: proposed mechanism of decomposition[J].AnalyticalChemistry, 2011, 83: 1069-1077.
[8] Zheng Z Y, Xu J C, Zhao J J. First-principles studies on the thermal decomposition behavior of FOX-7[J].HighPressureResearch, 2010, 30(2): 301-309.
[9] Frisch M, Trucks G, Schlegel H, et al. Gaussian 03[CP]//Revision E.01. Gaussian Inc. Wallingford, CT. 2004.
[10] 吴艳光, 罗运军, 葛震. GAP型交联改性双基推进剂黏合剂的力学性能[J]. 火炸药学报, 2012, 35(2): 66-69.
WU Yan-guang, LUO Yun-jun, GE Zhen. Mechanical. properties of the binder for GAP based cross-linked modified double-base propellant[J].ChineseJournalofExplosives&Propellants, 2012, 35(2): 66-69.
[11] 杨文升, 苟瑞君, 张树海, 等. HMX/NQ共晶分子间相互作用的密度泛函理论研究[J]. 火炸药学报, 2015, 38(6): 72-77
YANG Wen-sheng, GOU Rui-jun, ZHANG Shu-hai, et al. Study on theintermolecular interaction of HMX/NQ cocrystal explosive by density functional theory[J].ChineseJournalofExplosives&Propellants, 2015, 38(6): 72-77.
[12] Lu T, Chen F W. Multiwfn: a multifunctional wavefunction analyzer[J].JournalofComputationalChemistry, 2012, 33: 580-592.
[13] 肖鹤鸣, 朱卫华, 肖继军, 等. 含能材料感度判别理论研究——从分子、晶体到复合材料[J]. 含能材料, 2012, 20(5): 514-527.
XIAO He-ming, ZHU Wei-hua, XIAO Ji-jun, et al.Theoretical studies on sensitivity criterion of energetic materials—from molecules, crystals, to composite materials[J].ChineseJournalofEnergeticMaterials(HannengCailiao), 2012, 20(5): 514-527.
[14] 金朋刚, 常海, 陈智群, 等. FOX-7 的热分解动力学和机理研究[J]. 火工品, 2006 (2):20-24.
JIN Peng-gang, CHANG Hai, CHEN Zhi-qun, et al. Study on thethermal decomposition kinetics and mechanism of FOX-7[J].Initiators&Ptrotechnics, 2006 (2): 20-24.
[15] 宗和厚, 黄奕刚, 舒远杰, 等. FOX-7热分解起始机理及NO2对其催化效应的理论研究[J]. 含能材料, 2006, 14(6): 425-428.
ZONG He-hou, HUANG Yi-gang, SHU Yuan-jie, et al. Theoreticalstudy on the initial thermal decomposition and catalysis effects of NO2on FOX-7[J].ChineseJournalofEnergeticMaterials(HannengCailiao), 2006, 14(6): 425-428.
