一种四氢吡咯并吲哚化合物的合成及表征
2017-04-27董志兵王涛
董志兵,王涛
武汉工程大学化学与环境工程学院,湖北武汉 430205
一种四氢吡咯并吲哚化合物的合成及表征
董志兵,王涛
武汉工程大学化学与环境工程学院,湖北武汉 430205
四氢吡咯并吲哚骨架类生物碱由于其结构复杂性和显著的生物活性,引起了化学家、生物学家的广泛关注,同时,具有该核心单元(四氢吡咯并吲哚)的药物在临床治疗中也有一定的应用价值,因此,四氢吡咯并吲哚骨架的天然产物及其衍生物的合成近年来已成为药物化学研究领域的热点之一.报道了一种四氢吡咯并吲哚衍生物的合成途径:以色胺为起始原料,依次经过氨基的二碳酸二叔丁酯上保护、二氯二氰基苯醌氧、脱保护酸化、和吲哚乙酸缩合、在三氟乙酸中质子化加成,得到一种新的四氢吡咯并吲哚化合物,经过核磁共振氢谱和碳谱、高分辨质谱检测,确定了其结构.
四氢吡咯并吲哚;缩合;色胺;吲哚乙酸
哚类生物碱(见图1)广泛存在于动植物体体内,该类化合物具有重要的生理活性,如吗啡活性、抗癌活性和抗菌活性等[1-7].该类化合物的结构具有多样性,同时,为了获得更高的活性,化学家也努力对该类型化合物进行修饰.对该类化合物的衍生大多集中在吲哚吡咯环上,包括烷基化,芳基化或者手性控制等.大量的该类生物碱由两个吡咯吲哚结构组合而成[8-14].该类化合物的合成具有重要研究意义,长期以来,天然或者人工合成的各类吡咯并吲哚类化合物引起了合成化学家、生物学家的广泛关注.

图1 具有吲哚并吡咯结构的生物碱Fig.1Alkaloids with pyrroloindoline structure
本研究以色胺为起始物,合成了一种新的吲哚吡咯化合物,合成路线见图2.合成的关键步骤是最后一步:吲哚质子化环化反应,即酰胺的氮原子对吲哚2位的加成反应.该步合成中,摸索了几种酸催化反应的条件,最终发现:在三氟乙酸做溶剂时,反应才能充分进行,并且该步反应的产率高达95%.该化合物的合成路线如下图(图2)所示,各步的反应条件分别为:(a)二氧六环,(BOC)2O;(b)VTHF/VH2O=9∶1,DDQ;(c)TFA;(d)DMF,NaHCO3,EDCI,HOBT;(e)TFA.

图2 新型吲哚吡咯化合物的合成路线Fig.2Synthetic route of novel pyrroloindoline derivative
1 实验部分
1.1 仪器与试剂
所有底物都是来自Merck或Aldrich或Alfa Aesar,所有试剂都严格按照标准方法处理.核磁采用Varian-400型核磁共振仪(TMS为内标,CDCl3或DMSO-d6作溶剂),液质联用仪器采用Waters ACQUITY UPLC system液质联用色谱仪,高分辨质谱用Finnigan MAT 95Q质谱仪测定轰击源为EI;反应用TLC和LC-MS跟踪;分离方法为用柱色谱分离纯化,所用硅胶粒径为0.05 mm~0.75 mm.
1.2 N-叔丁氧羰基色胺(2)的合成
取色胺(2.0 g,12.48 mmol)于100 mL三口瓶中,依次加入50 mL二氧六环,二碳酸二叔丁酯(3.0 g,13.73 mmol),三乙胺(2.0 mL,14.1 mmol),磁力搅拌3 h,取出磁子,于30℃低压蒸除溶剂.随后加入20 mL乙酸乙酯溶解,水洗3次,再用饱和食盐水洗,硫酸钠干燥,粗产物用硅胶柱层析分离纯化(V石油醚∶V乙酸乙酯=2∶1)得白色固体N-叔丁氧羰基色胺∶白色固体(3.1 g),产率为95.38%.1H NMR(400 MHz,CDCl3):8.27(1H,s,1H,NH),7.62(1H,d,J=7.9Hz,ArH),7.38(1H,d,J=7.9 Hz,ArH),7.21-7.24(1H,m,ArH),7.13-7.16(1H,m,ArH),7.02(1H,s,ArH),4.67(1H,s,NH),3.48(2H,m,2-H),2.97(2H,t,J=6.7 Hz,2-H),1.47(9H,s,3-H).HRMS(ESI)m/z:calcd.For C20H17N3O2260.151 7,found 260.151 9.
1.3 化合物3的合成
将N-叔丁氧羰基色胺(2)(2.0 g,7.29 mmol)溶于20 mLVTHF/VH2O=9∶1(体积比)的溶剂中,缓慢滴加到溶于30 mL THF/H2O的DDQ(16.04 mmol,3.62 g)溶液中,反应混合物在室温下搅拌5 h,在减压下蒸出溶剂.反应混合物溶于50 mL乙酸乙酯溶剂中,用40 mL饱和的碳酸氢钠溶液洗,反复洗5次,用无水硫酸钠干燥.粗产物用硅胶柱层析分离纯化(V石油醚∶V乙酸乙酯=2∶1)得白色固体3(1.50 g),产率71.26%.1H NMR(400 MHz,DMSO-d6):12.01(1H,br s,NH),8.42(1H,d,J=3.0 Hz,ArH),8.17(1H,d,J=6.6 Hz,ArH),7.48(1H,dd,J=1.8 Hz,5.6 Hz,ArH),7.25-7.17(2H,m,ArH),7.02(1H,t,J=5.8 Hz,NH),4.30(2H,d,J=5.8 Hz,2-H),1.42(9H,s,(CH3)3C).HRMS(ESI)m/z:calcd.For C20H17N3O2274.131 7,found 274.131 5.
1.4 化合物4的合成
将化合物3(1.50 g,5.47 mmol)加入到15 mL三氟乙酸中,室温下搅拌2 h,减压蒸出溶剂三氟乙酸(溶剂用碳酸氢钠水溶液中和,废液pH值呈中性至弱碱性后被收集于废液桶),得到白色固体化合物(1.56 g),产率99.0%.1H NMR(400 MHz,DMSO-d6):12.51(1H,br s,NH),8.52(1H,d,J=3.4 Hz,ArH),8.41(3H,br s,NH2,-COOH),8.15-8.17(1H,m,ArH),7.52-7.54(1H,m,ArH),7.22-7.27(2H,m,ArH),4.36(2H,d,J=5.5 Hz,2-H).HRMS(ESI)m/z:calcd.For C12H11F3N2O3288.0722,found 288.0732.
1.5 化合物5的合成
取化合物4(1.0 g,3.47 mmol)于50 mL三口瓶中,加入DMF 25 mL,碳酸氢钠(0.7 g,8.33 mmol),EDCI[1-(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐,7 g,3.65 mmol],HOBT(1-羟基苯并三氮唑,0.5 g,3.70 mmol),吲哚乙酸(4.00 mmol)反应混合物在室温下搅拌12 h,减压蒸出溶剂,混合物溶于30 mL乙酸乙酯,用20 mL水洗3次,饱和食盐水洗,硫酸钠干燥.粗产物用硅胶柱层析分离纯化(V石油醚∶V乙酸乙酯=1∶1)得到白色固体5(0.56 g),产率为49.0%.1H NMR(400 MHz,DMSO-d6):11.98(1H,s,NH),10.98(1H,s,NH),8.40(1H,d,J=3.2 Hz,NH),8.09-8.16(2H,m,ArH),7.59(1H,d,J=8.0 Hz,ArH),7.47(1H,t,J=7.7 Hz,ArH),7.35(1H,d,J=7.8 Hz,ArH),7.27(1H,m,ArH),7.20(2H,m,ArH),7.07(1H,t,J=7.6 Hz,ArH),6.97(1H,t,J=7.7 Hz,ArH),4.47(2H,t,J=3.2 Hz,2-H),3.64(2H,s,2-H).HRMS(ESI)m/z:calcd.For C20H17N3O2331.131 5,found 331.131 3.
1.6 化合物6的合成
取化合物5(100 mg,0.30 mmol)于5 mL反应瓶中,加入3 mL三氟乙酸,反应混合物在室温下搅拌6 h,减压下蒸出溶剂三氟乙酸(溶剂用碳酸氢钠水溶液中和,废液pH值呈中性至弱碱性后被收集于废液桶),粗产物用硅胶柱层析分离纯化(V石油醚∶V乙酸乙酯=1∶3)得到白色固体(95 mg),产率为95.0%.1H NMR(400 MHz,DMSO-d6):12.21(1H,s,NH),8.46(1H,s,ArH),8.14(1H,d,J=7.9 Hz,ArH),7.48(1H,d,J=7.7 Hz,ArH),7.19(2H,m,ArH),7.09(1H,d,J=7.8 Hz,ArH),6.98(1H,t,J= 7.9 Hz,ArH),6.73(1H,s,NH),6.63(1H,t,J= 7.8 Hz),6.52(1H,d,J=7.9 Hz,ArH),5.42(1H,d,J=6.4 Hz,1-H),4.80(1H,d,J=14.1 Hz,2-H),4.46(1H,d,J=14.1 Hz,2-H),4.01(1H,m,2-H),2.93(1H,m,2-H).13C NMR(400 MHz,DMSO-d6):189.05,172.67,149.20,136.54,134.08,131.28,128.19,125.14,124.38,122.98,121.13,118.24,114.03,112.36,109.28,76.56,45.56,36.29,20.73.HRMS(ESI)m/z:calcd.ForC20H17N3O2331.131 5,found 331.131 3.
2 结果与讨论
以色胺为起始物,经过上保护、氧化、脱保护、缩合、质子化加成等五步反应合成出一种新的吲哚并吡咯结构的化合物[15],产物经过氢谱、碳谱、液质联用仪分析确定了结构.具有该结构的天然产物,大多具有良好的生物活性.该类化合物的合成能为具有吲哚并吡咯结构的天然产物的合成提供一个新的选择.另外,对化合物5的合成做了进一步分析,通过液质联用仪监测反应表明,出现两个副产物的峰,并且分子量和底物与两个吲哚乙酸缩合产生副产物分子量(M=488.1)吻合,推测一部分产物生成之后,产物吲哚氮继续与吲哚乙酸缩合,造成缩合反应的产率不高,如图3所示.
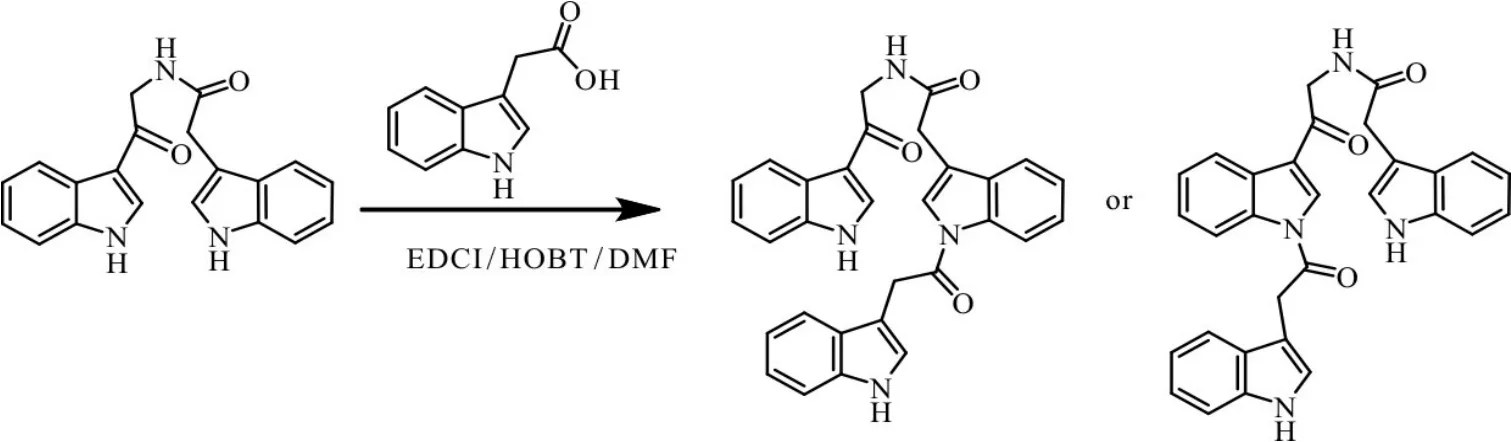
图3 缩合反应副产物分析Fig.3Analysis of byproducts of the condensation reaction
最终产物的合成最后一步是关键步骤:质子化加成反应,对该步反应条件进行了摸索,如图4所示.反应条件的摸索采用液质联用仪检测,对照实验显示:底物在醋酸中不反应;在以N,N-二甲基甲酰胺作溶剂条件下,催化量的硫酸催化不反应;对甲苯磺酸催化不反应;催化量的三氟乙酸能获得产物,但产率不到5%,但以三氟乙酸做溶剂的条件下,反应产率达到95%.

图4 质子化加成反应条件摸索Fig.4Exploration of reaction conditions of protonation-addition
3 结语
本文报道了一种四氢吡咯并吲哚的合成:以色胺为起始物,经过叔丁氧羰基保护、氧化、脱保护基、缩合、吡咯并吲哚环的构建等五步反应合成出新的四氢吡咯并吲哚化合物,总产率为31.32%.同时也对缩合反应的副产物和最终吡咯环的构建反应条件进行了摸索,发现以三氟乙酸做溶剂的条件下,质子化加成反应的产率很高,达到95%.本合成途径为构建四氢吡咯并吲哚单元的化合物提供了一种合成方法,显示出一定的应用价值.
[1]SMITH B P,TYLER M J,KANEKO T,et al.Evidence for biosynthesis of pseudophrynamine alkaloids by an Australian myobatrachid frog(Pseudophryne)and for sequestration of dietary pumiliotoxins[J].Journal of Natural Products,2002,65(4):439-447.
[2]TSUKAMOTO S,HIROTA H,KATO H,et al.Uro-chordamines A and B:larval settlement/metamorpho-sis-promoting,pteridine-containing physostigmine alka-loids from the tunicate Ciona savignyi[J].Tetrahe-dron Letters,1993,34(30):4819-4822.
[3]RUIZ-SANCHIS P,SAVINA S A,ALBERICIO F,et al.Structure,bioactivity and synthesisof natural products with hexahydropyrrolo[2,3-b]indole[J]. Chemistry-AEuropeanJournal,2011,17(5):1388-1408.
[4]HOLST P B,ANTHONI U,CHRISTPHRERSON C,et al.Marine alkaloids,15.Two alkaloids,flustramine E and debromoflustramine B,from the marine bryozoan Flustra foliacea[J].Journal of Natural Product,1994,57(7):997-1000.
[5]BROSSI A.Bioactive alkaloids.4.Results of recent investigations with colchicine and physostigmine[J]. Journal of Medicinal Chemistry,1990,33(9):2311-2319.
[6]GRIEG N H,PEI X F,SONCRANT T T,et al. Phenserine and ring C hetero-analogs:Drug candidates for the treatment of Alzheimer's disease[J].Medicinal Research Reviews,1995,15(1):3-31.
[7]SANO M,BELL K,MARDER K,et al.Safety and efficacyof oral physostigmineinthetreatment of Alzheimer disease[J].Neuropharmacol,1993,16(1):61-69.
[8]CAI Q,LIU C,LIANG X W,et al.Enantioselective construction of pyrroloindolines via chiral phosphoric acid catalyzed cascade Michael addition-cyclization of tryptamines[J].Organic Letters,2012,14(17):4588-4590.
[9]MASANORI T,KAZUHIRO H,TAKAMO I,et al. Thionium-basedone-potconstructionofhomo-/ heterodimericpyrroloindolinefromtryptamine[J]. Organic Letters,2014,16:3613-3615.
[10]REPKA L M,REISMAN S E.Recent developments in the catalytic,asymmetric construction of pyrroloin-dolines bearing all-carbon quaternary stereocenters[J]. Journal of Organic Chemistry.2013,78:12314-12320.
[11]AUSTIN J F,KIM S G,SINZ C J,et al.Enantioselective organocatalytic construction of pyrroloindolines by a cascadeaddition-cyclizationstrategy:synthesisof(-)-flustramine B[J].PNAS,2004,101(15):5482-5487.
[12]KIEFFER M E,CHUANG K V,REISMAN S E. Copper-catalyzeddiastereoselectivearylationof tryptophanderivatives:totalsynthesisof(+)-naseseazines A and B[J].Journal of the American Chemical Society,2013,135:5557-5560.
[13]LEE T B K,WONG G S K.Asymmetric alkylation of oxindoles:anapproachtothetotalsynthesisof(-)-physostigmine[J].Journal of Organic Chemistry,1991,56:872-875.
[14]STEVENA,OVERMMAN L E.Total synthesis of complexcyclotryptaminealkaloids:stereocontrolled construction of quaternary carbon stereocenters[J]. Angewandte Chemie,International Edition,2007,46:5488-5508.
[15]NICOLAOU K C,HUANGX H,LING T T,et al. Chemistry and biology of diazonamide A:first total synthesis and confirmation of the true structure[J]. Journal of the American Chemical Society,2004,126(14):12888-12896.
本文编辑:张瑞
Synthesis and Characterization of a Novel Pyrroloindoline Compound
DONG Zhibing,WANG Tao
School of Chemistry and Environmental Engineering,Wuhan Institute of Technology,Wuhan 430205,China
Alkaloids containing a pyrroloindoline unit attract extensive attention of chemists and biologists for their structural complexity and significant biological properties,and chemicals bearing pyrroloindoline skeleton were applied in clinical treatment.Thus,the synthesis of natural products bearing pyrroloindoline unit and their derivatives is a hot topic in medicinal chemistry for recent years.Hereby,we reported a synthetic method for the preparation of this novel compound which contains pyrroloindoline unit.Starting from tryptamine,then subsequently by di-tert-butyl pyrocarbonate protection of amino group,oxidation by using 2,3-dicyano-5,6-dichlorobenzoquinone,deprotection of the amino and acidification,condensation with indole-3-acetic acid,cyclization in trifluoroacetic acid,the desired product was furnished with good yield,and the structure of the target molecule is confirmed by1H NMR,13C NMR and high resolution mass spectrometry.
tetrahydropyrroloindoline;condensation;tryptamine;indole-3-acetic acid.
O621
A
10.3969/j.issn.1674-2869.2017.02.001
1674-2869(2017)02-0103-05
2017-01-03
国家自然科学基金(201302150);教育部留学回国人员基金[2012]1707;湖北省教育厅重点基金(D20131501);
武汉工程大学研究生创新基金(CX20130110);武汉工程大学校长基金(2014038)
董志兵,博士,教授.E-mail:zhibingdong80@aliyun.com
董志兵,王涛.一种四氢吡咯并吲哚化合物的合成及表征[J].武汉工程大学学报,2017,39(2):103-107.
DONG Z B,WANG T.Synthesis and characterization of a novel pyrroloindoline compound[J].Journal of Wuhan Institute of Technology,2017,39(2):103-107.
