非茂金属催化剂的合成及其聚合反应动力学
2017-04-19吴为果张艺镡王梦雪米普科
吴为果,张艺镡,王梦雪,米普科,许 胜
(华东理工大学 材料科学与工程学院 上海市先进聚合物材料重点实验室,上海 200237)
非茂金属催化剂的合成及其聚合反应动力学
吴为果,张艺镡,王梦雪,米普科,许 胜
(华东理工大学 材料科学与工程学院 上海市先进聚合物材料重点实验室,上海 200237)
以配体2,2’-硫代(4,6-二氯苯酚)与不同的钛化合物合成了4种均相非茂金属催化剂,利用1H NMR,MS,13C NMR等方法分析了配体、催化剂以及聚合产物的结构,通过非茂金属催化剂催化乙烯聚合测定了聚合反应体系的相关动力学参数,考察了温度和压力对聚合反应速率的影响。实验结果表明,以倍半乙基氯化铝为助催化剂催化乙烯聚合时,聚合反应速率很高。催化剂Ⅰ(钛酸四丁酯与2,2’-硫代(4,6-二氯苯酚)合成)制得的聚乙烯中的支链含量很低,约占聚乙烯的0.103%。催化剂Ⅰ的动力学曲线为缓升平稳型,聚合反应级数为1.099,接近1级反应。随聚合温度的升高,聚合反应速率逐渐增大。非茂金属催化剂催化乙烯聚合反应的表观活化能为37.49 kJ/mol。
非茂金属催化剂;倍半乙基氯化铝;乙烯聚合;聚合动力学
非茂金属催化剂是继茂金属催化剂之后烯烃聚合领域兴起的又一种高效催化剂[1]。由于催化剂的特性高度依赖金属活性中心周围的空间结构和电子效应,因此在对催化剂设计的过程中,主要从增加金属活性中心的电子云密度和稳定性方面进行考虑。对非茂金属催化剂的研究主要集中在催化剂结构与聚合产物性能的关系上。如何将催化剂的空间结构和电子效应更好地结合,使催化剂在聚合反应过程中发挥最优化作用,是非茂金属催化剂的主要研究方向及关键。
研究催化剂的聚合反应动力学对于优化聚合反应工艺条件具有现实的指导意义,同时有助于开发新型催化剂体系。目前,对Ziegler-Natta催化体系[2-3]和茂金属催化体系[4-5]聚合反应动力学的研究已有很多报道,但非茂金属催化体系因研究时间较短,相关的聚合反应动力学研究相对较少。
本工作以配体2,2’-硫代(4,6-二氯苯酚)与不同的钛化合物合成了4种均相非茂金属催化剂,利用1H NMR,MS,13C NMR等方法分析了配体、催化剂以及聚合产物的结构,通过非茂金属催化剂催化乙烯聚合测定了聚合反应体系的相关动力学参数,考察了温度和压力对聚合反应速率的影响。
1 实验部分
1.1 主要试剂
倍半乙基氯化铝(EASC)、甲基铝氧烷(MAO)、四氯化钛、钛酸四丁酯、钛酸异丙酯:分析纯,阿拉丁化学有限公司;2,2’-硫代(4,6-二氯苯酚):分析纯,梯希爱(上海)化成工业发展有限公司。对空气敏感或水敏感物质的操作都在高纯氩气(纯度99.9%)保护下使用标准Schlenk 技术进行。溶剂使用前在氩气保护和金属钠存在下加热回流24 h后使用。
1.2 催化剂的制备
将配体2,2’-硫代(4,6-二氯苯酚)和不同的钛化合物分别溶于甲苯中;在100 mL的Schlenk瓶中,将2,2’-硫代(4,6-二氯苯酚)甲苯溶液缓慢滴加入钛化合物甲苯溶液中,得到橙黄色溶液;升温回流3 h后降至一定温度,恒温搅拌反应12 h;减压蒸馏将反应产生的异丙氧醇和甲苯与非茂金属配合物分离;分离后的固体经溶剂洗涤和干燥得橙黄色固体粉末,即为非茂金属催化剂。
分别采用钛酸四丁酯、钛酸异丙酯和四氯化钛与2,2’-硫代(4,6-二氯苯酚)反应得到催化剂记为催化剂Ⅰ~Ⅲ,增加2,2’-硫代(4,6-二氯苯酚)的用量再与钛酸四丁酯反应得到的催化剂记为催化剂Ⅳ。催化剂Ⅰ~Ⅳ的合成路线见式(1)~(4),结构见图1。
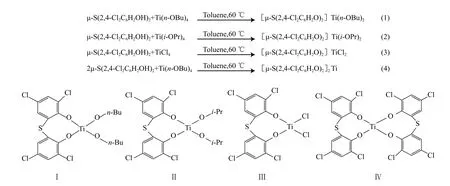
图1 催化剂Ⅰ~Ⅳ的结构Fig.1 Structures of catalystⅠ-Ⅳ.
1.3 乙烯聚合
将250 mL高压反应釜分别用高纯氩气置换3次、纯化后的乙烯置换3次,然后升至预定温度,称取计算量的非茂金属催化剂、100 mL甲苯及助催化剂EASC,通过加料器加入干燥无氧的高压反应釜中,再引入乙烯至预定压力后进行聚合。加入10%(w)的盐酸乙醇溶液终止反应,用乙醇和水洗至中性,烘干至恒重,得到白色粉末为聚乙烯。
1.4 测试及表征方法
1H NMR表征采用瑞士布鲁克公司Bruker AVANCE-400型核磁共振仪,CDCl3为溶剂,四甲基硅烷为内标;MS表征采用HP公司HP5989A型质谱仪采用EI方式在70 eV下测定;高温13C NMR表征采用布鲁克公司AVANCE Ⅲ 400 MHz型核磁共振仪测定,氘代邻二氯苯为溶剂,测试温度135 ℃;聚乙烯的黏均相对分子质量以十氢萘为溶剂,在135 ℃恒温硅油浴中,用乌氏黏度计测定。
2 结果与讨论
2.1 配体及催化剂的表征结果
[μ-S(2,4-Cl2C6H2OH)2]的1H NMR(CDCl3,400 MHz)表征结果为:化学位移δ = 6.95~7.28(4H,Ph),5.79(2H,Ph-OH)。
[μ-S(2,4-Cl2C6H2O)2]Ti(O-nBu)2的1H NMR(CDCl3,400 MHz)表征结果为:δ = 6.95~7.28(4H,Ph),1.56(4H,—CH2—),1.38(4H,—CH2—),1.24(12H,—CH3);MS (70 eV)表征结果为:m/z = 418.8(61,M+—nBu—nOBu),474.9(100,M+—nOBu),504.9(15,M+CH3CH2CH3),547.9(26,M+)。
[μ-S(2,4-Cl2C6H2O)2]Ti(O-iPr)2的1H NMR (CDCl3,400 MHz)表征结果为:δ = 7.09~7.19(4H,Ph),3.64(2H,—OCH—),1.17(12H,—CH3);MS(70 eV)表征结果为:m/z = 418.8(91,M+-iPr-iOPr),460.9(71,M+CH2CH2CH2CH3),504.9(34,M+CH3),519.9(33,M+)。
[μ-S(2,4-Cl2C6H2O)2]2Ti的1H NMR(CDCl3,400 MHz)表征结果为:δ = 7.01~7.12(8H,Ph);MS(70 eV)表征结果为:m/z = 418.8(89,M+S(2,4-Cl2C6H2)2O),460.9(65,M+S(2,4-Cl2C6H2)2),504.9(34,M+CH3),755.7(38,M+)。
2.2 乙烯聚合结果
不同助催化剂对非茂金属催化剂聚合的影响见表1。由表1可知,以AlEt3和MAO为助催化剂时,聚合反应速率很低,而以EASC为助催化剂时,聚合体系表现出很高的聚合反应速率,达2.55×106g/(mol·h),这归因于EASC在溶液中产生了Et2AlCl和EtAlCl2,可使阳离子稳定,并将金属中心Ti还原生成具有高催化活性的Ti-烷基中间体[6-7]。催化剂的电子效应和位阻因素对聚合性能的影响也不同;催化剂Ⅰ~Ⅲ的聚合反应速率依次降低,这是因为异丙氧基和正丁氧基为供电子基团,Cl为吸电子基团,供电效应使金属活性中心周围的电子云密度增大。催化剂Ⅲ的金属活性中心的电子云密度较低,而催化剂Ⅰ和Ⅱ的空间位阻较大,大位阻可防止活性物种因失去配体而分解,还能使阳离子活性中心与平衡的阴离子更加离散,活性中心将有一个更加开放的配位空间,使金属活性中心更加稳定,聚合反应速率增加[8-9]。

表1 不同助催化剂对非茂金属催化剂聚合反应的影响Table 1 Effects of different co-catalysts on the ethylene polymerization with the non-metallocen
催化剂Ⅰ制得的聚乙烯的13C NMR谱图见图2。从图2可看出,δ= 132.9处的峰归属于聚乙烯链端亚甲基;δ = 127.3~130.8处的峰归属于顺式和反式的碳碳双键。催化剂Ⅰ的聚合反应机理见图3。从图3可看出,非茂金属催化剂的空间位阻大,在一定聚合压力下容易发生β-H链转移,在生成末端带乙烯基的聚合物链的同时形成新的活性中心,并继续催化乙烯单体进行聚合,因此聚合反应可一直保持较高的聚合反应速率。

图2 催化剂Ⅰ制得的聚乙烯的13C NMR谱图Fig.213C NMR spectrum of the polyethylene prepared with catalyst Ⅰ.

图3 催化剂Ⅰ的聚合反应机理Fig.3 Mechanism of the polymerization with catalystⅠ.
用Lindeman-Adams方法[10]得到聚合物13C NMR谱图中各峰的化学位移,再按文献[11]报道的方法得到聚合物支链的含量。计算结果表明,催化剂Ⅰ制得的聚乙烯中的支链含量很低,约占聚乙烯的0.103%,其中,甲基支链含量为0.057%、乙基支链含量为0.046%。这是因为在一定压力下,端自由基从非端基碳上夺取氢原子后,非端基碳变成新自由基并在此增长,从而在聚合物链上形成支链。
3 聚合反应动力学
3.1 聚合反应动力学曲线
研究表明,烯烃聚合反应动力学曲线一般可分为缓升平稳型(A)、速升缓降型(B)、缓解型(C)和高活性速降型[12](D)(见图4a)。催化剂Ⅰ制得的聚乙烯的黏均相对分子质量随聚合时间的变化见表2。根据表2得到的聚合反应速率随时间变化的曲线见图4b。由图4可知,催化剂Ⅰ的动力学曲线为缓升平稳型,聚合反应速率在反应60 min前一直处于缓慢上升期,60 min时达到最大,然后聚合反应速率有较小幅的衰减并保持相对稳定。

表2 催化剂Ⅰ制得的聚乙烯黏均相对分子质量随聚合时间的变化Table 2 Relationship between Mvand polymerization time

图4 常见聚合反应动力学曲线(a)[12]和催化剂Ⅰ的聚合反应动力学曲线(b)Fig.4 Common polymerization kinetics curves(a)[12]and the kinetic curve of the ethylene polymerization with catalystⅠ(b).
3.2 聚合反应速率方程
聚合反应速率(Rp)与单体浓度(cM)通常呈1级依赖关系[13],即Rp∝cM。而Kissin等[14]研究报道了聚合反应速率与单体浓度呈非1级依赖关系,分别为1.24和1.8。Chien等[15]在研究茂金属催化体系反应动力学时发现,聚合反应速率对单体浓度的非1级反应级数依赖性是因为单体和活性位之间形成了配合物。虽然烯烃聚合动力学的研究结果存在各种差异,但基本动力学规律没有改变。
如果反应体系的聚合活性高,链引发反应消耗的乙烯单体量可忽略,则聚合反应速率可表示为:

式中,Kp为链增长反应速率常数,L/(mol·h);Ktr为链转移反应速率常数,L/(mol·h);c*为活性中心浓度,g/L;cM为乙烯单体浓度,g/L;t为聚合反应时间,s;n为反应级数。
由于该非茂金属催化体系的聚合速率很大,即Kp>Ktr,链转移消耗的乙烯单体量也可忽略,则式(5)可变为:

聚合反应速率随时间的延长而逐渐增大,一段时间后保持基本不变,即达到稳态,则Kp和c*均为常数,将式(6)两边取对数得到:

不同压力和温度下乙烯单体在甲苯中的溶解度[16]见图5。
由图5可知,当温度为100 ℃、压力在0~5 MPa范围内时,乙烯压力与甲苯溶液中的乙烯含量基本呈线性关系,该温度下乙烯压力(p)与甲苯溶液中乙烯摩尔含量(y)的关系见式(8)。

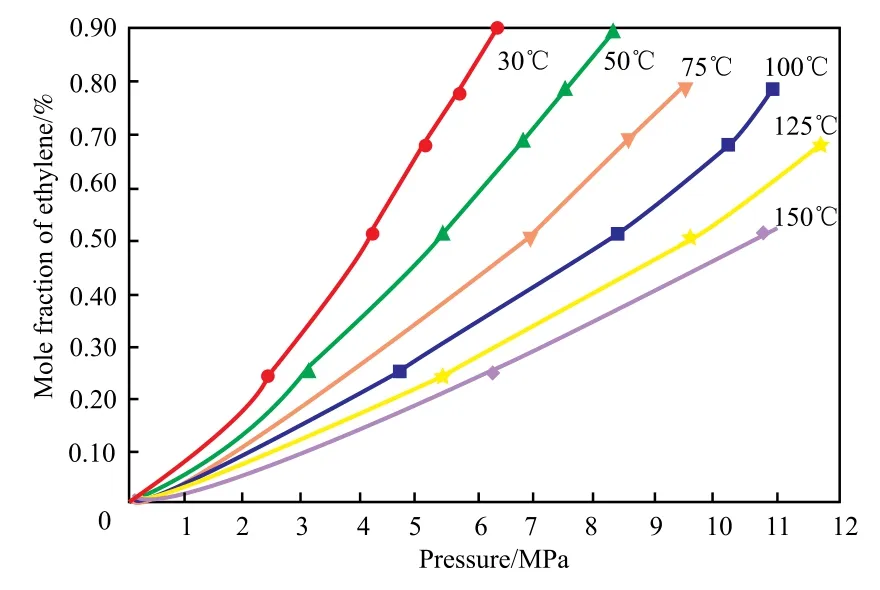
图5 不同压力和温度下乙烯在甲苯溶液中的溶解度Fig.5 Solubility of ethylene in toluene solution under different pressure and temperature.
100 ℃、不同乙烯压力下的乙烯单体浓度和聚合反应速率见表3。

表3 不同乙烯压力下的乙烯单体浓度和聚合反应速率Table 3 The rate of polymerization and the concentration of ethylene monomer under different ethylene pressure
由式(7)可知,以lnRp对lncM做图,可由斜率求得反应级数n(见图6)。

图6 lnRp~lncM关系图Fig.6 lnRpvs. lncM.
从图6可求得lnKpc*= 11.196 6,斜率n = 1.099,接近1级反应,故聚合反应速率可表示为:

3.3 反应温度的影响
一般情况下,非茂前过渡金属催化体系的热稳定性较茂金属催化剂高[17],因为非茂金属催化剂的供电效应及空间位阻能提高金属活性中心的电子云密度,增加催化剂的热稳定性[18]。升高聚合温度虽会使乙烯单体在甲苯中的溶解度降低,但也让催化剂更容易被引发,且有利于活性中心-助催化剂EASC复合物(紧密离子对)与自由态活性中心的平衡偏向于后者,乙烯单体与活性中心接触的机会增加,从而使乙烯插入过程更加容易,聚合反应速率增加;但超过一定温度后,聚合反应速率随温度升高而降低,因为高温高压下配体和金属中心脱离,造成活性物种分解,加速催化剂的失活[19],降低烯烃在反应体系中的溶解度,使聚合反应速率降低;此外,随聚合温度的升高会使β-H链转移反应速率加快,导致聚乙烯相对分子质量降低[20]。不同聚合温度下的单体浓度和聚合反应速率见表4。

表4 不同温度下的单体浓度和聚合反应速率Table 4 The concentration of ethylene monomer and the rate of polymerization at different temperature
由表4可看出,温度在60~100 ℃范围内,随聚合温度的升高,乙烯单体在甲苯溶液中溶解度逐渐降低,而聚合反应速率呈逐渐增大的趋势,表明聚合温度的升高使链引发反应加快,并且活性中心与氯离子形成更为松散的离子对,从而使乙烯单体更容易插入金属—碳键进行链增长反应,使聚合反应速率增加。
表观速率常数与聚合温度的关系一般符合Arrhenius公式,故有:

式中,Kp0为链增长反应速率常数,L/(mol·h);R为理想气体常数,8.314 J/(mol·K);Ea为表观活化能,kJ/mol;T为温度,K。由式(9)可得到:

结合式(10)和式(11)得到:

将式(12)两边求对数得到:


图7 不同聚合温度下的ln(Rp/cM1.099)~1/TFig.7 ln(Rp/cM1.099) vs. 1/T.
4 结论
1)以配体2,2’-硫代(4,6-二氯苯酚)与不同的钛化合物合成了均相非茂金属催化剂Ⅰ~Ⅳ。以EASC为助催化剂催化乙烯聚合时,非茂金属催化体系表现出很高的聚合反应速率。钛酸四丁酯与2,2’-硫代(4,6-二氯苯酚)合成的催化剂Ⅰ制得的聚乙烯中的支链含量很低,约占聚乙烯的0.103%。
2) 催化剂Ⅰ的动力学曲线为缓升平稳型。聚合反应级数为1.099,接近1级反应,聚合反应速率Rp可以表示为
3) 随聚合温度升高,聚合反应速率逐渐增大。根据Arrhenius公式求得非茂金属催化剂催化乙烯聚合反应的表观活化能为37.49 kJ/mol。
[1] Gibson V C,Spitzmesser S K. Advances in non-metallocene olefin polymerization catalysis[J].Chem Rev,2003,103(1):283-315.
[2] Marques M M,Dias A R,Costa C,et al. Homogeneous Ziegler-Natta polymerization:Akinetic approach:Ⅰ. Steadystate kinetics[J].Polym Int,1997,43(1):77-85.
[3] Marques M M,Dias A R,Justino J,et al. Homogeneous Ziegler-Natta polymerisation:Akinetic approach:Ⅱ. Transient-state kinetics[J].Polym Int,1997,43(1):86-96.
[4] Yuan Youling,Tao Ruoyuan,Wang li,et al. Kinetics of ethylene polymerization catalyzed by metallocene[J].J Mol Catal,2002,16(2):92-96.
[5] Mi Puke,Xu Sheng,Liu min,et al. Study on kinetics of ethylene polymerization catalyzed by supported binuclear metallocene[J].J Mol Catal,2012,26(6):537-545.
[6] Umare P S,Tiwari A J,Antony R,et al. Synthesis of ultralow-molecular-weight polyethylene wax using a bulky Ti(Ⅳ)aryloxide-alkyl aluminum catalytic system[J].Appl Organomet Chem,2007,21(8):652-660.
[7] Umare P S,Antony R,Gopalakrishnan K,et al. Synthesis of low molecular weight polyethylene waxes by a titanium BINOLate-ethylaluminum sesquichloride catalyst system[J].J Mol Catal A:Chem,2005,242(1/2):141-150.
[8] Matsui S,Mitani M,Saito J,et al. Post-metallocenes:Catalytic performance of new bis(salicylaldiminato) zirconium complexes for ethylene polymerization[J].Chem Lett,2000(5):554-555.
[9] Matsui S,Mitani M,Saito J,et al. A family of zirconium complexes having two phenoxy-imine chelate ligands for olefin polymerization[J].J Am Chem Soc,2001,123(28):6847-6856.
[10] Lindeman L P,Adama J Q. Carbon-13 nuclear magnetic resonance spectrometry:Chemical shifts for the paraffins through C9[J].Anal Chem,1971,43(10):1245-1252.
[11] Galland G B,de Souza R F,Mauler R S,et al.13C NMR determination of the composition of linear low-density polyethylene obtained with [η3-methallyl-nickel-diimine]PF6 complex[J].Macromolecules,1999,32(5):1620-1625.
[12] Keii T,Soga K. Catalytic polymerization of olefins[M].Amsterdam:Elsevier Science Ltd,1989:35-36.
[13] ChakravartiS,Ray W H. Kinetic study of olefin polymerization with a supported metallocene catalyst:Ⅲ. Ethylene homopolymerization in slurry[J].J Appl Polym Sci,2001,81(12):2901-2917.
[14] Kissin Y V,Mink R I,Nowlin T E,et al. Kinetics and mechanism of ethylene homopolymerization and copolymerization reactions with heterogeneous Ti-based Ziegler-Natta catalysts[J].Top Catal,1999,7(1/4):69-88.
[15] Chien J C,Yu Zhengtian,Marques M M,et al. Polymerizations of olefins and diolefins catalyzed by monocyclopentadienyltitanium complexes containing a (dimethylamino)ethyl substituent and comparison with ansa-zirconocene systems[J]. J Polym Sci Part A:Polym Chem,1998,36(2):319-328.
[16] Miller S A. Ethylene and its industrial derivatives[M].London:Ernest Benn Limited,1969:16-17,22,632-635.
[17] Sumitomo Chemical Company. Catalyst for olefin polymerization and process for producing olefin polymer using the catalyst:US 5280000[P].1994-01-18.
[18] Matsui S,Tohi Y,Mitani M,et al. New bis(salicylaldiminato) titanium complexes for ethylene polymerization[J]. Chem Lett,1999(10):1065-1066.
[19] Hu Jie. Organometallic olefin polymerization catalysts and polyolefins[M].Beijing:Chemical Industry Press (CIP),2010:105-106.
[20] Svejda S A,Brookhart M. Ethylene oligomerization and propylene dimerization using cationic (α-diimine)nickel(Ⅱ) catalysts[J].Organometallics,1999,18(1):65-74.
[21] Roos P,Meier G B,Samson J J C,et al. Gas-phase polymerization of ethylene with a silica supported metallocene catalyst:Influence of temperature and deactivation[J].Macromol Rapid Commun,1997,18(4):319-324.
(编辑 邓晓音)
Synthesis of non-metallocene catalysts and their polymerization kinetics
Wu Weiguo,Zhang Yixin,Wang Mengxue,Mi Puke,Xu Sheng
(Shanghai Key Laboratory of Advanced Polymeric Materials,School of Materials Science and Engineering,East China University of Science and Technology,Shanghai 200237,China)
A series of homogeneous non-metallocene catalysts were synthesized from titanium compounds and 2,2’-thiobis(4,6-dichlorophenol) as ligands. The kinetics of ethylene polymerization with the catalysts was investigated and the kinetics parameters were obtained. The ligands,catalysts and obtained polymers were characterized by means of1H NMR,MS and13C NMR. The effects of pressure and temperature on the polymerization were discussed. It was showed that,when ethylaluminum sesquichloride was used as the co-catalyst,the reaction rate of the ethylene polymerization was very high. The branching degree of the obtained polymers was very low about 0.103%. The kinetics curve of catalystⅠwas slowly and stably rising,and the polymerization reaction order was 1.099,approaching a pseudo first order reaction. With the polymerization temperature rise,the polymerization rate increased gradually. The apparent activation energy of the polymerization with the non-metallocene catalysts was 37.49 kJ/mol.
non-metallocene catalysts;ethylaluminum sesquichloride;ethylene polymerization;polymerization kinetics
1000-8144(2017)02-0183-07
TQ 426.92
A
10.3969/j.issn.1000-8144.2017.02.007
2016-07-19;[修改稿日期]2016-11-28。
吴为果(1989—),男,湖南省浏阳市人,硕士生,电邮 smiledoors@126.com。联系人:米普科,电邮 pkmi@ecust.edu.cn。
中国石油科技创新基金项目(2014D-5006-0504)。
