施氮与不施氮条件下玉米开花期相关性状的 QTL 定位
2017-04-14郭向阳陈建军卫晓轶祝云芳王安贵刘鹏飞汤继华陈泽辉
郭向阳,陈建军,卫晓轶,祝云芳,王安贵,刘鹏飞,汤继华,陈泽辉*
(1 贵州省旱粮研究所,贵阳 550006;2 贵州省遵义市辉煌种业有限责任公司,遵义 563100;3 河南农业大学农学院,郑州 450002;4 河南省新乡市农业科学院,新乡 453000)
施氮与不施氮条件下玉米开花期相关性状的 QTL 定位
郭向阳1,陈建军2,卫晓轶3,4,祝云芳1,王安贵1,刘鹏飞1,汤继华3*,陈泽辉1*
(1 贵州省旱粮研究所,贵阳 550006;2 贵州省遵义市辉煌种业有限责任公司,遵义 563100;3 河南农业大学农学院,郑州 450002;4 河南省新乡市农业科学院,新乡 453000)
【目的】探究施氮与不施氮对玉米开花期相关性状变异的影响,并定位相关 QTLs。 【方法】以玉米的骨干自交系综 3 为供体亲本,许 178 为受体亲本,通过杂交、回交和分子标记辅助选择的方法,构建了一套以许178 为遗传背景的综 3 染色体单片段代换系 (SSSLs) 群体,其中包含 160 个单片段代换系。以这套 SSSLs 以及许 178 为材料,采用裂区试验设计,在施氮 (N+) 和不施氮 (N-) 条件下,通过一年三点 (贵州贵阳、德江和云南罗平) 的表型评价,利用复合区间作图法对玉米散粉期、吐丝期和散粉-吐丝间隔期 (ASI) 3 个开花期相关性状进行 QTL 定位。 【结果】在基因组范围内,2 种氮处理条件下共定位到 54 个开花期相关性状 QTLs,主要定位在第 1、3、6、9 和 10 染色体上。其中 5 个 QTLs 在 3 个环境中均被检测到。施氮条件下,吐丝期 (DTS) 相关位点 qDTS9a,位于第 9 染色体,可解释表型变异的 3.05%;吐丝散粉间隔期 (ASI) 相关位点 qASI10a,位于第10 染色体,可解释表型变异的 30.74%。不施氮条件下,散粉期 (DTP) 相关位点 qDTP9,可解释表型变异的3.43%;吐丝期相关位点 qDTS9a,位于第 9 染色体,可解释表型变异的 4.08%;ASI 相关位点 qASI10,位于第10 染色体,可解释表型变异的 50.28%。对不同处理条件下的定位结果比较发现,不施氮条件下检测到的 QTLs数目显著高于施氮条件下的检测数量。 【结论】不同氮素处理下存在一些共有的控制玉米开花期相关的遗传区段,分别位于 Bin9.02 (umc1170-umc1636-bnlg1401-umc1271) 和 Bin10.04 (umc1077-umc1053-umc2350),这些区段可能在玉米氮素吸收、转运和利用过程中起重要作用,可作为下一步精细定位和图位克隆玉米开花期相关基因的重要候选区域。
玉米;SSSL;不同氮处理;开花期相关性状;数量性状基因座 (QTLs)
氮作为玉米必需的大量营养元素之一,在其生长和发育过程中起着重要作用。长期以来,为追求玉米产量,氮肥被大量使用在玉米的栽培过程中。有报道显示,发达国家的农业氮肥使用量超过 500万吨,发展中国家则远远超过该数字。过量使用氮肥 造 成 土 壤 、 淡 水 以 及 大 气 等 严 重 污 染[1]。 长 期 以来,为探索玉米产业可持续发展途径,研究者从土壤供氮能力、施肥量、作物本身的氮素吸收和利用能力等方面对氮素利用效率进行了大量的探索,揭示了不同地区的施肥量与玉米高产潜力的关系,说明了不同土壤的供氮能力对玉米产量提高的作用,并在此基础上培育出一系列的高产、高效优势玉米自交系或杂交种。如 Mo17,掖 478 等在低氮条件下往往表现出严重的缺氮症状,而大多数自交系表现为中间类型。沈 125 表现出一定程度的低氮耐受性[2]。刘建安等[3]研究表明,不同杂交种对氮水平的反应存在显著差异。刘宗华等[4]利用许 178 和黄 C 构建的F2∶3群体研究了不同氮水平条件下玉米株高变异的遗传差异,结果表明许 178 具有较黄 C 更高的低氮耐受性,并定位到了一些与氮素利用效率相关的重要QTLs。郑祖平等[5]利用 Mo17 和黄早四构建的 RIL 群体 定 位 到42 个 可 能 与 玉 米 对 低 氮 胁 迫 相 关 的QTLs。然而,氮素吸收利用是一个复杂的网络调控过程,涉及到很多微效位点或基因,且不同性状在不同氮处理条件下,其遗传差异也不尽相同。因此,针对不同性状,构建相应的分离群体并以不同的表型性状作为目标,探究不同氮处理水平对目标表型变异的影响并定位出相应的遗传位点,将会为深入解析玉米氮素利用效率的遗传基础提供进一步的支持。
开花期作为玉米重要农艺性状,对玉米籽粒产量、地域与季节的适应性都具有十分重要的决定作用[6]。不同来源种质之间的开花期相关性状存在丰富的变异,在温带材料与热带种质之间的差异尤其突出。为研究不同种质开花期变异的遗传基础,研究者基于不同群体,定位了大量与开花期相关遗传位点。目前,在 MaizeGDB 上已经公布的玉米花期相关 QTLs 达 201 个(http://www.maizegdb.org/qtl.php),其中 156 个 QTLs 与散粉期、吐丝期和开花-吐丝间隔期 (ASI) 直接相关[7]。前人利用不同群体对玉米开花期相关性状进行了定位研究;Veldboom 等[8]利用Mol7 × H99 的 F2∶3家系,将影响散粉期的 QTL 定位到 1、5、6、7、8 和 9 染色体上;影响吐丝期的 5个 QTLs 定位在 1、2、6、7 和 8 染色体上,影响ASI 的 QTLs 定位在第 6 和 8 染色体上。Sari-Gorla等[9]利用 B73 × H99 的 RILs 群体和 Ribaut 等[10]利用Ac7643 × Ac7729/TZSRW 的 F2∶3家系,定位到的与散粉期、吐丝期、ASI 相关的 QTLs 与前者有所差异。此外,Agrama 等[11]、Khairallah 等[12]、Austin 等[13]也对玉米花期相关性状进行了 QTL 定位。兰进好[6]研究表明,花期性状 QTL 的基因作用方式以加性和部分显性为主,上位性效应对玉米花期性状具有十分 重 要 的 作 用 , 韩 娅 楠 等[14]研 究 也 得 出 同 样 结 果 。Li 等[15]利用黄早四 × 掖 107 的 F2∶3群体,在灌溉和干旱胁迫下,分别检测到 3 个和 2 个控制 ASI 的QTLs,分别位于第 1、2、3 和第 2、5 染色体;同时,发现 ASI 与产量性状 QTL 存在显著关系。胡彦民等[16]利用琼 68 和 K12 的 F2∶3群体定位到 22 个开花期相关的 QTLs,散粉期 5 个、吐丝期 4 个、散粉至吐丝间隔期 5 个、散粉周期 4 个、吐丝周期 2个、花丝活力 2 个,结果发现多数 QTL 存在环境表达的特异性。Gonzalo 等[17]利用 B73 和 Mo17 构建的RILs 群体,对不同密度下的开花期相关性状进行定位,分别在第 2、8 染色体上检测到开花期相关QTLs。但是利用开花期相关性状作为目标,探究不同氮素处理对其表型变异的影响并定位相关 QTLs 的研究则少有报道。以单片段代换系为材料,采用施氮和不施氮两种处理,鉴定不同施氮水平下的开花期相关性状变异情况,并利用符合区间作图法定位控制花期相关性状的 QTLs,借助不同处理条件下的定位结果比较,鉴定出两种条件下的共有位点。通过对两种施氮条件下的玉米开花期相关性状进行QTL 定位,发掘控制玉米开花期相关的遗传区段,借助不同条件下的定位结果一致性分析,鉴定出与氮素吸收利用显著相关的候选遗传区段,为揭示玉米氮素效率利用差异以及分子辅助的提高玉米氮利用效率育种提供分子遗传学依据。
1 材料与方法
1.1 供试材料
本研究所用的 160 份玉米单片段代换系由河南农业大学汤继华教授提供。该群体是以优良玉米自交系综 3 为供体亲本、许 178 为受体亲本,通过杂交、回交、自交,结合 SSR 分子标记辅助选择的方法构建而成。综 3 和许 178 作为中国玉米育种中的骨干材料,在生产上发挥了重要作用,以这两个材料为亲本之一的重要玉米杂交种如豫玉 22 号 (综 3 × 87 - 1) 和农大 108 (黄 C × 许 178) 等在生产上被大面积推广应用。
1.2 试验区概况与试验设计
2015 年分别在贵州省德江县 (北纬 28°15′51″,东经 108°06′39″)、贵阳市 (北纬 26°34′13″,东经106°42′39″) 和云南省罗平县 (北纬 24°31′,东经103°57′) 进行田间试验。试验采用裂区设计,不同氮处理水平作为主区,设施氮 (N+) 和不施氮 (N-) 两个水平,三个亲本群体 (SSSLs) 作为副区。单行区,小区行长 4.0 m,宽行 0.7 m,每行 17 株,株距0.235 m,密度为 52500 plant/hm2,每个处理 2 次重复。对代表花期相关性状的散粉期、吐丝期和散粉-吐丝间隔期进行鉴定和检测。散粉期是指播种至全区 50% 以上植株雄花开始散粉的天数,吐丝期是指播种至全区 50% 以上植株雌穗花丝吐出的天数,散粉-吐丝间隔是指从散粉期到吐丝期之间的天数。试验点各小区供试土壤耕层养分含量见表 1。
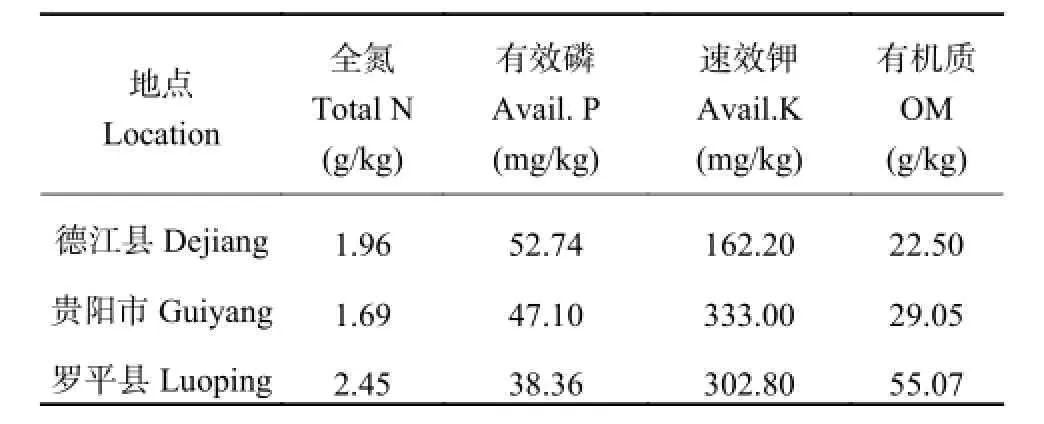
表1 0—20 cm 土层基础肥力状况Table 1 Soil fertility at the 0-20 cm layer
施氮处理在苗期和大喇叭口期各施尿素375 kg/hm2(N+) 。 每 个 小 区 均 基 施 过 磷 酸 钙375 kg/hm2、硫酸钾 187.5 kg/hm2,田间管理措施同大田。
1.3 表型数据分析与 QTL 定位
采用 SPSS17.0 统计软件,对 160 份 SSSLs 的开花期进行单因素的方差分析和 Duncan’s 多重比较。本研究中涉及到的基因型鉴定数据的相关研究结果[18]已发表。利用完备区间作图法对生育期相关 QTL 进行定位,若某一性状与许 178 在 P < 0.05 水平上差异显著,则认为该单片段代换系代换片段上存在一个 QTL,并同时估算各个 QTL 的加性效应值及贡献率[19]。
加性效应值 (A) = (单片段代换系的表型值 - 对照的表型值)/2;
贡献率 (R) = 加性效应值/对照的表型值 × 100%。
2 结果与分析
2.1 开花期相关性状
不同 SSSLs 开花期相关性状的表型调查结果列于表 2。施氮条件下,散粉期不同亲本群体在德江的变异范围为 84.5~94.5 d,平均值为 88.3 ± 1.9 d;在贵 阳 和 罗 平 的 变 异 范 围 分 别 为74.0~86.0 d 、68.0~80.5 d。吐丝期不同 SSSLs 在德江、贵阳和罗平的变异范围分别为 90.0~99.5 d、76.5~87.0 d 和71.0~84.0 d。散粉-吐丝间隔期 (ASI) 不同 SSSLs 在德江、贵阳和罗平的变异范围分别为 1.5~6.5 d、1.0~5.0 d 和 1.0~6.5 d。
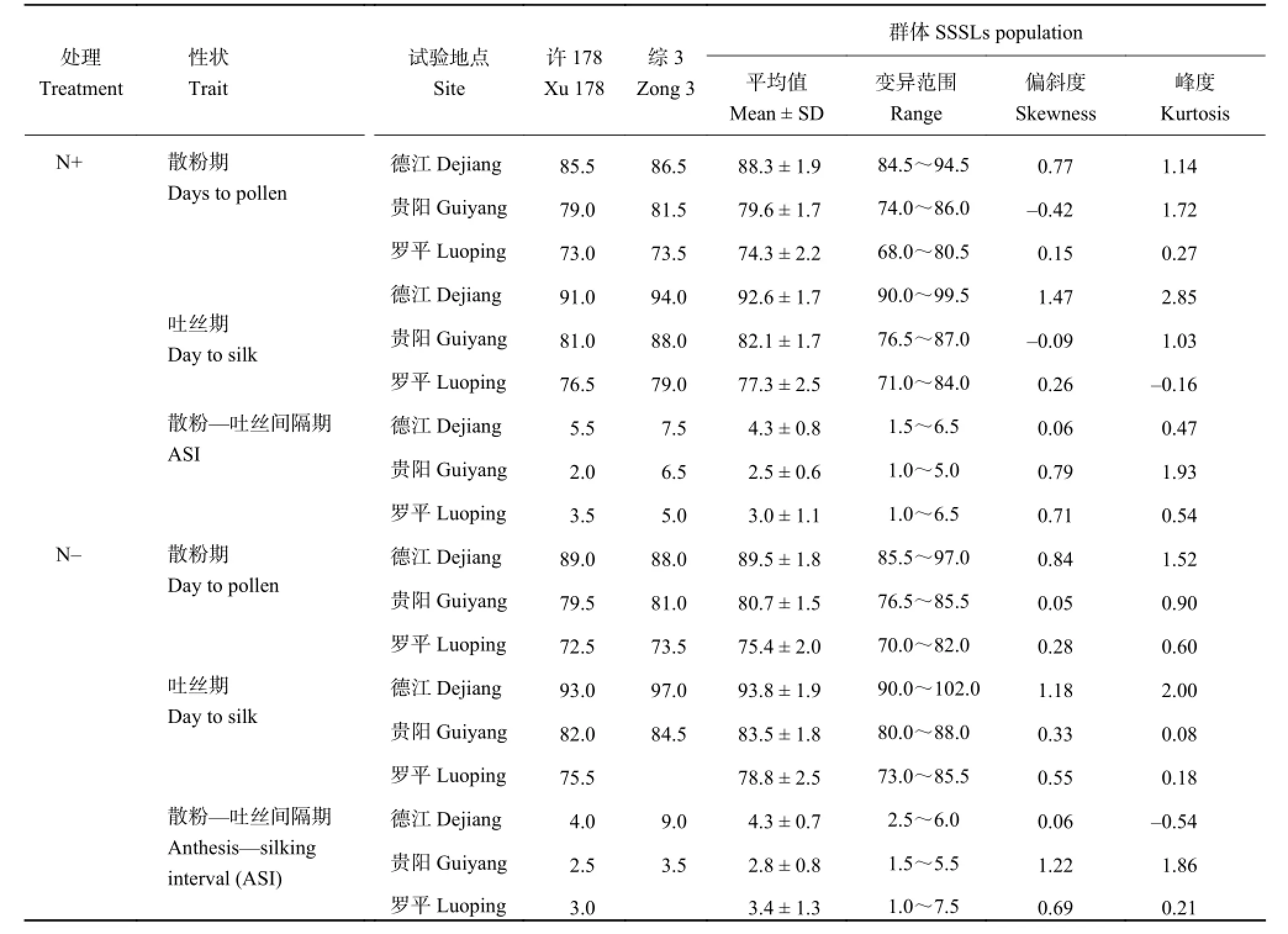
表2 SSSLs 开花期相关性状表型Table 2 Performance of SSSLs population in the three experimental sites
不施氮条件下,不同 SSSLs 在德江、贵阳和罗平 的 散 粉 期 的 变 化 范 围 分 别 为 85.5~ 97.0 d、76.5~85.5 d 和 70.0~82.0 d;吐丝期的变化范围分别为 90.0~102.0 d、80.0~88.0 d 和 73.0~85.5 d;ASI 变 化 范 围 分 别 为2.5~6.0 d、1.5~5.5 d 和1.0~7.5 d。开花期相关性状在不同处理条件下均表现出较为丰富的表型变异,为后续的 QTL 定位提供了较为丰富的表型数据。
2.2 控制玉米开花期相关性状的 QTLs
对不同氮素处理条件下的开花期性状 QTL 定位结果列于表 3。结果显示,施氮 (N+) 和不施氮 (N-)两 种 处 理 条 件 下 , 共 检 测 到 54 个 开 花 期 性 状QTLs,分布在除第 2 和 4 染色体外的其它染色体上。其中在德江试验点,两种氮处理水平下共检测到 18 个 QTLs;在贵阳试验点,两种氮处理水平下共检测到 17 个 QTLs;在罗平试验点,两种氮处理水平下共检测到 19 个 QTLs;其中单个 QTL 可解释目标表型变异的变幅为 2.2%~80.0%。
在施氮条件下,3 个地点共鉴定出 6 个控制散粉期的一致性 QTLs,分布于第 1、6、7、10 染色体上,单个 QTL 可解释的表型变异介于 2.5%~8.8%之间,其中 1 个 QTLqDTP6 在两个地点 (德江、罗平) 被同时检测到,位于 Bin6.04,在德江和罗平点的贡献率分别为 8.8% 和 3.4%,且来自于许 178 的等位基因起增效作用。对于吐丝期,在 3 个环境条件下共鉴定到 7 个 QTLs,分布在第 1、3、5、6、7、9 染色体上,单个 QTL 所解释的表型变异介于2.5%~7.7% 之间。其中 1 个 QTLqDTS9a 在三个地点被同时检测到,位于 Bin9.0 区域,其贡献率分别为 2.5% (德江)、3.1% (贵阳) 和 3.6% (罗平),且来自许 178 的等位基因起增效作用。对于 ASI,在 3 个地点共鉴定到 4 个共有 QTLs,分布在第 5、9 和 10 染色体上,单个 QTL 所解释的表型变异介于 13.6%~62.5% 之间。1 个 QTLqASI10a 在三个地点被同时检测到,位于 Bin10.04 区域;其贡献率分别为 13.6% (德江)、50.0% (贵阳) 和 28.6% (罗平),来自于许 178的等位基因起增效作用。
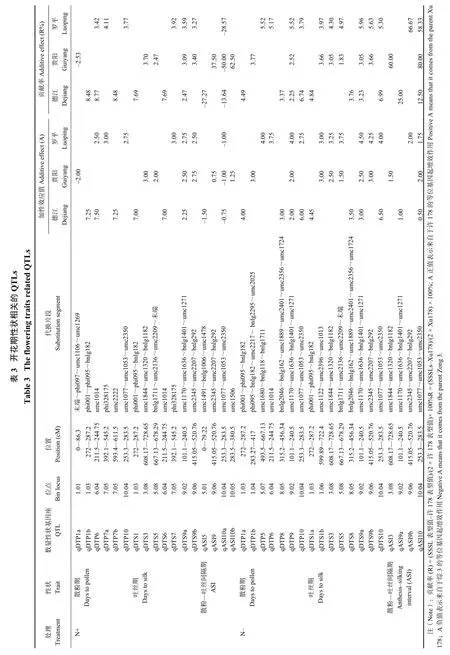
不施氮条件下,3 个地点共鉴定到 7 个散粉期相关 QTLs,分布在第 1、5、6、8、9 和 10 染色体上,单个 QTL 所解释的表型变异介于 2.2%~6.7%之间。1 个 QTLqDTP9 在三个地点被同时检测到,位于 Bin9.02,可解释的表型变异分别为 2.2% (德江)、2.5% (贵阳) 和 5.5% (罗平),来自于许 178 的等位基因起增效作用。在 3 个地点共鉴定到 8 个吐丝期 QTL,分布在第 1、3、5、8、9、10 染色体上,单个 QTL 所解释的表型变异介于 1.8%~7.0% 之间。1 个 QTLqDTS9a 在三个地点被同时检测到,位Bin9.02,在 3 种环境的贡献率分别为 3.2% (德江)、3.0% (贵阳) 和 6.0% (罗平),来自于许 178 的等位基因起增效作用。在 3 个地点共鉴定到 4 个散粉-吐丝间隔期 QTL,分布在第 3、9 和 10 染色体上,单个QTL 所解释的表型变异介于 12.5%~80.0% 之间,1个 QTLqASI10a 在三个地点被同时检测到;qASI10a位于 Bin10.04,在 3 种环境的贡献率分别为 12.5% (德江)、80.0% (贵阳) 和 58.3% (罗平),来自于许 178的等位基因起增效作用。这些染色体区段将作为下一步精细定位和图位克隆玉米生育期相关性状候选基因的重点区域。
3 讨论
关于玉米氮高效利用的 QTL 定位研究前人已有报道[20-24]。Gallais 等[25]在高氮水平下和低氮水平下检测到的 QTL 有很大差别;Agrama 等[21]的研究发现在低氮和高氮水平下既有相同的 QTL 也有特异表达的QTL。本研究中,施氮条件下,在染色体 Bin1.03 区域,检测散粉期 QTLqDTP1b 和吐丝期 QTLqDTS1,代换片段均为 phi001-phi095-bnlg182,该研究结果与前人的定位结果存在一定的一致性,只是在他们的研究中,该区域存在一个分别与株型性状和丝黑穗病抗性相关的 QTL-qDH12[26-27],一定程度上反映出生育期与生物量以及抗病性之间是存在一定关系的。在 Bin6.04 区域,存在一个与散粉期和吐丝期显著相关的 QTLs (qDTP6 和 qDTS6),代换片段均为umc1014。在 Bin7.05 区域,检测到控制散粉期变异的 QTLs,qDTP7a 和 qDTP7b,吐丝期 QTLqDTS7,而前期报道则在该区域检测到一个与株高相关 QTL和叶面积相关 QTL qLA-6[26]。说明了在不同氮素处理条件下,一些共有的和特异性遗传位点共同起用,从而影响玉米氮素利用的效率。
除此之外,两种氮处理条件下,共检测到 31 个控制玉米开花期相关性状的 QTLs。不施氮条件下,在 染 色 体bin8.05 区 域 , 检 测 到2 个 散 粉 期QTLqDTP8 和 QTLqDTS8,其代换片断为 bnlg2046-bnlg162-umc1889-umc2401-umc2356-umc1724,在该区 域 存 在 穗 行 数 相 关 QTL qKRN8-2QTL[28]。 在Bin9.06 区 域 , 检 测 到1 个 吐 丝 期 相 关 QTL qDTS9b,代换片段为 umc2345-umc2207-bnlg292;在染色体 Bin10.04 区域,检测到 1 个吐丝期 QTL qDTS10,代换片段为 umc1077-umc1053-umc2350;与Khairallah 等[29]基于 F2 群体的定位结果较为一致,他们在该区域定位了一个控制 ASI 的 QTL;另外,Ducrocq 等[30]基于不同日照长短自交系构建的近等基因系群体,通过关联分析的方法也在该区域定位到一个与玉米散粉期相关的主效 QTLs,其贡献率为40%。进一步说明了本研究结果的可靠性。更为重要的是,本研究在两种氮处理条件下,共检测出在 3个试验环境中均稳定表达且效应较大的一致性QTLs,分别位于 Bin9.02 区域 (qDTS9a)(umc1170~umc1636~bnlg1401~umc1271) 和 Bin10.04 区域(qASI10a)(umc1077~umc1053~umc2350)。尽管这些区域在前人的研究中与穗粗、穗行数等显著相关[27],但是关于该群段在玉米氮素吸收、转运以及利用过程中的重要性的研究则没有。本研究结果证明了这些区段在玉米不同氮素处理过程中均起着重要作用,对于深入挖掘玉米氮素利用效率将提供更为可靠的分子遗传学信息。
4 结论
玉米在不同氮素处理条件下既存在一些共有的主效 QTL,如 qDTS9a、qASI10 在施氮和不施氮条件下均被检测到;同时还存在一些特异性的 QTL 区段,如 qDTP9 仅在不施氮条件下被检测到。这些染色体区段将为进一步改良玉米氮素利用效率以及相关候选基因的图位克隆等提供更多的分子遗传学支持。
致谢:贵州省旱粮研究所杨明伦老师在田间管理过程中给予帮助;贵州省旱粮研究所吴迅博士在文章修改中提出宝贵意见与建议;在此表示感谢。
[1]Ranmos C. Effect of agricultural practices on the nitrogen losses in the environment [J]. Nutrient Cycling in Agroecosystems, 1995, 43(1): 183-189.
[2]曹敏建, 衣莹, 佟占昌, 等. 耐低氮胁迫玉米的筛选 与评价 [J]. 玉米科学, 2000, 8 (4): 64-69. Cao M J, Yi Y, Tong Z C, et al. Indexes and comprehensive evaluation of low nitrogen tolerance of maize [J]. Journal of Maize Sciences, 2000, 8(4): 64-69.
[3]刘 建安, 米国华, 张福锁. 不 同基因型玉米 氮 效率差异的比 较 研究[J]. 农业生物技术学报, 1999, 7 (3): 248-254. Liu J A, Mi G H, Zhang F S. Difference in nitrogen efficiency among maize genotypes[J]. Journal of Agricultural Biotechnology, 1999, 7(3): 248-254.
[4]刘宗华, 汤继华, 王春丽, 等. 氮胁迫与非胁迫条件下玉米不同时期株高的动态QTL定位 [J]. 作物学报, 2007, 33 (5): 782-789. Liu Z H, Tang J H, Wang C L, et al. QTL analysis of plant height under N-stress and N-input at different stages in maize [J]. Acta Agronomica Sinica, 2007, 33 (5): 782-789.
[5]郑祖平, 黄玉碧, 田孟良, 等. 不同供氮水平下玉米株型相关性状的QTLs定位和上位性效应分析 [J]. 玉米科学, 2007, 15 (2): 16-18. Zheng Z P, Huang Y B, Tian M L, et al. Mapping QTLs and epistasis for plant type traits in maize under two nitrogen levels[J]. Journal of Maize Sciences, 2007, 15(2): 16-18.
[6]兰进好. 玉米开花期相关性状的QTL分析 [J]. 西北植物学报, 2010, 30(3): 471-480. Lan J H. QTL analysis on the flowering related traits in maize [J]. Acta Botanica Boreali-Occidentalia Sinica, 2010, 30 (3): 471-480.
[7]李贤唐, 丁俊强, 王瑞霞, 等. 玉米株型相关性状的QTL定位与分析[J]. 江苏农业科学, 2011, 39 (2): 21-25. Li X T, Ding J Q, Wang R X, et al. Maize plant type traits QTL mapping and analysis[J]. Jiangsu Agricultural Sciences, 2011, 39 (2):21-25.
[8]Velboom L R, Lee M. Molecular-marker-facilitated studies of morphological traits in maize II. Determination of QTLs for grain yield and yield components [J]. Theoretical and Applied Genetics, 1994, 89 (4): 451-458.
[9]Sari-Goria M, Krajewski P, Di Fonzo N, et al. Genetic analysis of drough tolerance in maize by molecular markers. II. Plant heigh and flowering [J]. Theoretical and Applied Genetics, 1999, 99: 289-295.
[10]Ribaut J, Hoisingtond, Deutseh J A. Identification of quantitative trait loci under drought conditions in tropical maize.2. Flowering parameters and the anthesis-silking interval [J]. Theoretical and Applied Genetics, 1996, 92: 905-914.
[11]Agrama H A, Moussa M E. Mapping QTLs in breeding for drought tolerance in maize (Zea mays L.) [J]. Euphytica, 1996, 91: 89-97.
[12]Khairallah M M, Bohn M, Jiang C, et al. Molecular mapping of QTL for southwestern corn borer resistance, plant height and flowering in tropical maize[J]. Plant Breeding, 1998, 117(4): 309-318.
[13]Austin D F, Lee M. Genetic resolution and verification of quantitative trait loci for flowering and plant height with recombinant inbred lines of maize [J]. Genome, 1996, 39: 957-968.
[14]韩娅楠, 刘福建, 王瑞霞, 等.玉米生育期QTL定位及上位性互作效应的遗传研究[J]. 华北农学报, 2010, 25 (2): 84-87. Han Y N, Liu F J, Wang R X, et al. QTL mapping and epistasis analysis of flowering related traits in maize [J]. Acta Agriculturae Boreali-Sinica, 2010, 25(2): 84-87.
[15]Li X H, Liu X D, Li M S, et al. Identification of quantitativer trait loci for anthesis-silking interval and yield components under drought stress in maize [J]. Acta Botanica Sinica, 2003, 45(7): 852-857.
[16]胡彦民, 吴欣, 李翠香, 等. 玉米制种花期相关性状的QTL分析 [J].南京农业大学学报, 2008, 31(1): 11-16. Hu Y M, Wu X, Li C X, et al. Genetic analysis on the related traits of florescence for hybrid seed production in maize [J]. Journal of Nanjing Agricultural University, 2008, 31 (1): 11-16.
[17]Gonzalo M, Holland J B, Vyn T J, et al. Direct mapping of density response in a population of B73×Mo17 recombinant inbred lines of maize (Zea Mays L.) [J]. Heredity, 2010, 104: 583-599.
[18]曹浩飞, 王彬, 毛克举, 等. 利用单片段代换系对玉米开花期相关性状的QTL定位 [J]. 河南农业大学学报, 2014, 48 (1): 6-10. Cao H F, Wang B, Mao K J, et al. Mapping of the QTL for flowering related traits in maize using a series of single segment substitution lines [J]. Tournal of Henan Agricultural University, 2014, 48 (1):6-10.
[19]Guo X, Guo Y P, Ma J, et al. Mapping heterotic loci for yield and agronomic traits using chromosome segment introgression lines in cotton[J]. Journal of Integrative Plant Biology, 2013, 55 (8):759-774.
[20]Lance R V, Lee M. Genetic mapping of quantitative trait loci in maize in stress and nonstress environments: Ⅰ. Grain yield and yield components[J]. Crop Science, 1996, 36: 1310-1319.
[21]Agrama H A S, Zakaria A G, Said F B, Tuinstra M. Identification of quantitative trait loci for nitrogen use efficiency in maize [J]. Molecular Breeding, 1999, 5: 187-195.
[22]Hirel B, Pascal B, Isabelle Q, et al. Towards a better understanding of the genetic and physiological basis for nitrogen use efficiency in maize [J]. Plant Physiology, 2001, 125(3): 1258-1270.
[23]Olivier L, Sylvain C, Patricia M, et al. Quantitative trait Loci analysis of nitrogen use efficiency in Arabidopsis [J]. Plant Physiology, 2003, 131: 345-358.
[24]Coque M, Gallais A. Genomic regions involved in response to grain yield selection at high and low nitrogen fertilization in maize [J]. Theoretical and Applied Genetics, 2006, 112: 1205-1220.
[25]Gallais A, Hirel B. An approach to the genetics of nitrogen use efficiency in maize [J]. Journal of Experimental Botany, 2004, 55(396): 295-306.
[26]许诚, 王彬, 毛克举, 等. 利用单片段代换系群体定位玉米株型性状QTL [J]. 玉米科学, 2014, 22 (2): 28-34. Xu C, Wang B, Mao K J, et al. QTL mapping for plant-type related traits using single segment substitution lines in maize [J]. Journal of Maize Sciences, 2014, 22(2): 28-34.
[27]张书红, 张世煌, 李新海, 等. 玉米抗病基因一致性图谱的构建 [J].中国农学通报, 2007, 23 (6): 601-606. Zhang S H, Zhang S H, Li X H, et al. Construction of consensus map of gene in maize [J]. Chinese Agricultural Science Bulletin, 2007, 23(6): 601-606.
[28]焦付超, 李永祥, 陈林, 等. 特异玉米种质四路糯的穗行数遗传解析[J]. 中国农业科学, 2014, 47(7): 1256-1264. Jiao F C, Li Y X , Chen L, et al. Genetic dissection for kernel row number in the specific maize germplasm four-rowed waxy corn [J]. Scientia Agricultura Sinica, 2014, 47(7): 1256-1264.
[29]Khairallah M M, Bohn M, Jiang C, et al. Molecular mapping of QTL for southwestern corn borer resistance, plant height and flowering in tropical maize [J]. Plant Breeding, 1998, 117(4): 309-318.
[30]Ducrocq S, Giauffret C, Madur D, et al. Fine mapping and haplotype structure analysis of a major flowering time quantitative trait locus on maize chromosome 10 [J]. Genetics, 2009, 183: 1555-1563.
QTL mapping of flowering related traits of maize with and without nitrogen application
GUO Xiang-yang1, CHEN Jian-jun2, WEI Xiao-yi3,4, ZHU Yun-fang1, WANG An-gui1,
LIU Peng-fei1, TANG Ji-hua3*, CHEN Ze-hui1*
( 1 Guizhou Institute of Upland Crops, Guiyang 550006, China; 2 Huihuang Seed Industry Co., LTD, Zunyi Guizhou 563100, China; 3 College of Agronomy, Henan Agricultural University, Zhengzhou 450002, China; 4 Xinxiang Academy of Agricultural Sciences of Henan Province, Xinxiang Henan 453000, China )
【Objectives】The purpose of this study was to dissect variation of maize flowering-related traits under two conditions of nitrogen applied normally or none, and to map the related QTLs. 【Methods】 One panel included 160 Single Segment Substitution Lines (SSSLs) was constructed by using foundation parental lines of Zong3 and Xu178 as donor and receptor parents, respectively. The panel was treated with two conditions of nitrogen applied normally or none by using the split-plot experiment design, and the nitrogen treatment was set as the main plot and genotypes were set as the sub-plots under three environments, including Guiyang, Dejiang in Guizhou Province and Luoping in Yunnan Province. Three flowering-related traits, including day to pollen (DTP),day to silk (DTS) and anthesis-silking interval (ASI), were investigated in one treatment. 【Results】The results showed that across the whole genome, about 54 flowering-related traits QTLs were identified under the two N treatments, which were mainly located on the chromosome 1, 3, 6, 9 and 10, among them, 3 QTLs were common under three environments. Under N+treatment, QTL qDTS9a controlling day to silling (DTS) was found which was located on chromosome 9, and could explain 3.05% genetic variation; another QTL qASI10a controlling the ASI was also found, which located on chromosome 10, and could explain 30.74% genetic variation. Under N-conditions, QTL qDTP9 was identified, which could explain 3.43% genetic variation of DTP; qDTS9a related with DTS was found, which located on chromosome 9, and could explain 4.08% genetic variation; QTL qASI10 was also identified, which located on chromosome 10, and could explain 50.28% genetic variation. Comparing between N+ and N- conditions showed that, the QTLs identified under N- was much more than those under N+ treatment. 【Conclusions】Two commonly QTLs are existed in the two nitrogen treatments, one is qDTS9a, which is located on Bin9.02, with flank markers of umc1170-umc1636-bnlg1401-umc1271, and the other is qASI10a, which is located on Bin 10.04, with flank markers of umc1077-umc1053-umc2350. These loci may play important role in nitrogen absorbing, transporting, and utilization during maize development, and served as candidate loci in further map-based cloning of maize flowering-related traits.
maize; single segment substitution lines (SSSLs); nitrogen levels; flowering related traits; quantitative trait locus (QTLs)
2016-07-18 接受日期:2016-11-22
贵州省农业科学院自主创新项目(黔农科院自主创新科研专项字[2014]006号);贵州省科技支撑计划项目(黔科合支撑[2016]2605号);国家“七大作物育种”专项(2016YFD0101206-4)资助。
郭向阳(1982—),男,河南滑县人,博士研究生,副研究员,主要从事玉米遗传育种研究。E-mail:xyguo0372@163.com
* 通信作者 E-mail:Tangjihua1@163.com;Tel:0851-83760096,E-mail:chenzh907@sohu.com
