坏死性凋亡介导化学性缺氧引起的HT22海马神经元损伤和炎症
2017-04-14侯娇艳周忠群田绍文
王 波,徐 勇,李 祥,侯娇艳,周忠群,田绍文,旷 昕
(南华大学 1.附属第一医院麻醉科、2.附属第一医院疼痛科、3. 医学院生理教研室,湖南 衡阳 421001)
坏死性凋亡介导化学性缺氧引起的HT22海马神经元损伤和炎症
王 波1,徐 勇1,李 祥1,侯娇艳1,周忠群2,田绍文3,旷 昕1
(南华大学 1.附属第一医院麻醉科、2.附属第一医院疼痛科、3. 医学院生理教研室,湖南 衡阳 421001)
目的 探讨坏死性凋亡(necroptosis)是否参与化学性缺氧诱导的小鼠HT22海马细胞损伤和炎症。方法 采用化学性缺氧模拟剂氯化钴(CoCl2)作用HT22细胞建立化学性缺氧损伤的细胞模型。蛋白质免疫印迹法测定受体相互作用蛋白3(receptor interacting protein 3,RIP3)的水平;应用细胞计数试剂盒(cell counting kit-8,CCK-8)测定海马细胞的存活率;乳酸脱氢酶(lactate dehydrogenase,LDH)试剂盒检测细胞培养液中的LDH活性;罗丹明123染色荧光显微镜照相法检测线粒体膜电位(mitochondrial membrane potential, MMP);双氯荧光素染色荧光显微镜照相法测定胞内活性氧(reactive oxygen species,ROS)生成水平;ELISA法检测细胞培养液中白介素-1β(IL-1β)和肿瘤坏死因子-α(TNF-α)的水平。结果 600 μmol·L-1CoCl2作用HT22细胞36 h可产生明显的细胞毒性作用,使细胞存活率降至(52.0±2.65)%,成功建立化学性缺氧损伤的海马细胞模型;此外,CoCl2可引起HT22细胞的多种损伤和炎症,表现为培养液中LDH活性升高,ROS过度生成,MMP丢失以及炎症因子(IL-1β和TNF-α)分泌均增多。40~100 μmol·L-1坏死性凋亡抑制剂necrostatin-1(Nec-1)共处理可抑制CoCl2引起的HT22细胞存活率降低,其中80 μmol·L-1时对细胞毒性的抑制作用最明显。同时,80 μmol·L-1Nec-1可对抗CoCl2可引起HT22细胞的上述多种损伤和炎症。此外,CoCl2处理HT22细胞6~48 h可促进RIP3的表达水平,Nec-1可明显抑制CoCl2对RIP3表达的上调作用。结论 坏死性凋亡介导化学性缺氧诱导的HT22海马神经元损伤和炎症。
坏死性凋亡;化学性缺氧;氯化钴;HT22细胞;损伤;炎症
缺氧是机体最常见的应激之一,是导致神经元损伤的一种重要的病理生理机制,因此,深入探讨缺氧的神经元损伤机制对防治神经损伤具有重要的理论意义和临床指导价值。研究表明[1-3],低氧损伤神经细胞的机制是多方面的,如细胞毒性作用、活性氧(reactive oxygen species,ROS)过度生成(氧化应激)[2-4]、线粒体损伤[2-4]、内质网应激[5]、炎症反应[6]和激活某些信号分子通路,如丝裂原激活蛋白激酶(mitogen-activated protein kinase,MAPKs)[2,4]、核因子-κB(nuclear factor-κB,NF-κB)[5]、Akt[也称蛋白激酶B(protein kinases,PKB)][7]等。此外,缺氧也可引起神经细胞的死亡,其死亡方式包括凋亡(apoptosis)[1-4]、坏死(necrosis)[8]、自噬(autophagy)[9]和坏死性凋亡(necroptosis)[10-12]。坏死性凋亡是Degterev等[10]于2005年在小鼠脑缺血模型中发现的,其具有坏死的细胞形态学特征,并和凋亡一样具有严格的程序性调控机制,且有特异性的抑制剂necrostatin-1(Nec-1),因此这种死亡方式也被称为程序性坏死(programmed necrosis)[11]。受体相互作用蛋白1(receptor interacting protein 1,RIP1)和受体相互作用蛋白3(receptor interacting protein3,RIP3)的活化并形成复合物是启动坏死性凋亡的关键,其中RIP3被视为坏死性凋亡的特异性检测指标,RIP3的表达增多提示坏死性凋亡的发生,下调或沉默RIP3的表达可不同程度地阻止坏死性凋亡[10-12]。近年来,多项研究表明,神经缺血缺氧损伤可引起坏死性凋亡发生。Kelliher等[13]报道,在小鼠大脑中动脉闭塞(middle cerebral artery occlusion, MCAO)的实验模型中,应用坏死性凋亡的抑制剂Nec-1可减少脑组织梗死面积,提示脑梗死可引起坏死性凋亡发生。Vieira等[14]证实,海马神经元细胞经氧葡萄糖剥夺(oxygen glucose deprivation,OGD)处理后,RIP1和RIP3表达增多,Nec-1不仅能减少RIP3的表达,还能减少OGD引起的海马神经元死亡。然而,坏死性凋亡在缺氧引起的海马神经元损伤和炎症中的作用,目前尚未完全清楚。
为此,本研究应用化学性缺氧模拟剂氯化钴(cobalt chloride,CoCl2)损伤小鼠HT22海马细胞建立化学性缺氧的神经细胞损伤模型,着重探讨坏死性凋亡在化学性缺氧引起的海马细胞损伤和炎症中的作用,为进一步阐明缺氧诱导海马神经元损伤的机制提供新颖的实验依据。
1 材料与方法
1.1 材料 抗RIP3抗体来源于Cell Signaling(USA);罗丹明123(Rhodamine 123,Rh 123)、CoCl2、双氯荧光素( 2’,7’-dichlorfluorescein-diacetate,DCFH-DA)、Nec-1、乳酸脱氢酶(lactate dehydrogenase,LDH)检测试剂盒购自Sigma-Aldrich公司(USA);特级胎牛血清(fetal bovine serum,FBS)购自Gibco BRL(USA);细胞计数试剂盒8(cell counter kit-8, CCK-8)购自Dojindo Lab(Japan);DMEM培养基由Hyclone公司(USA)供应;IL-1β和TNF-α ELISA试剂盒由武汉华美生物工程有限公司提供。HT22海马细胞取自于中山大学实验动物中心细胞库。
1.2 细胞培养及实验分组 HT22海马细胞于含5% CO2的37 ℃恒温培养箱中培养,生长在含10% FBS的DMEM培养基中,当细胞生长至约80%的融合状态时可进行实验。实验分为4组:(1) Control组:DMEM培养基作用海马细胞36 h;(2) CoCl2组:600 μmol·L-1CoCl2作用海马细胞36 h;(3)Nec-1+ CoCl2组:80 μmol·L-1Nec-1和600 μmol·L-1CoCl2共处理海马细胞36 h;(4) Nec-1组:80 μmol·L-1Nec-1与DMEM共处理海马细胞36 h。
1.3 RIP3蛋白表达测定 海马细胞在60 mm培养皿中生长至约80%融合度时,给予不同的处理后加入细胞裂解液作用30 min后,11 400×g高速离心10 min,对上清液采用BCA法进行蛋白质含量。等量蛋白经十二烷基硫酸钠聚丙烯酰胺凝胶(SDS-PAGE)电泳分离后,转移至PVDF膜上,经5%脱脂奶粉封闭1 h后加入抗RIP3或GAPDH抗体(浓度均为1 ∶1000),4 ℃作用过夜后加入II 抗稀释液孵育1.5 h。ECL法使PVDF膜显色,暗室中曝光到X线片上,凝胶成像扫描系统分析结果。实验重复3次。
1.4 细胞存活率测定 在96孔板中种植海马细胞,按照分组分别处理后,每孔加入CCK-8 溶液10 μL,培养箱内孵育2.5 h,酶标仪上读取450 nm处的吸光度值(A)。细胞存活率按照以下公式计算:细胞存活率/%=处理组A /对照组A×100%。实验重复3次。
1.5 LDH活性测定 海马细胞在96孔板中生长至融合度约80%时,按照设定的分组处理后,留取培养液作为检测标本,按照LDH试剂盒说明书操作,检测并计算出培养液中LDH的活性。实验重复3次。
1.6 细胞内ROS水平测定 海马细胞在24孔板中生长至融合度约80%时,按照分组分别处理后,加入DCFH-DA染液后于37 ℃温箱中作用30 min,荧光显微镜(TE-2000 Nikon,日本)下随机照片记录5个高倍镜视野,应用图像分析软件(Image J 1.47i)软件计算出绿色荧光的强度的平均值[平均荧光强度(mean fluorescent intensity,MFI),其数值大小能间接反映ROS水平的高低],并对每组数据进行统计分析。实验重复3次。
1.7 线粒体膜电位(mitochondrial membrane potential,MMP)水平测定 海马细胞在24孔板中生长至约80%的融合度时,按照分组给予相应的处理后,加入Rh 123 缓冲液,37 ℃温箱中作用30 min,荧光显微镜下随机照片记录5个高倍镜视野,应用图像分析软件计算出绿色荧光的MFI(数值大小可反映MMP的高低),并对每组数据进行统计分析。实验重复3次。
1.8 炎症因子分泌水平测定 海马细胞在96孔板中生长至融合度约80%时,按照分组给予不同处理后,收集培养基作待测标本,按照ELISA试剂盒说明书进行操作,测定出细胞培养液中IL-1β和TNF-α的水平。实验重复3次。
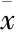
2 结果
2.1 CoCl2诱导HT22海马细胞化学性缺氧损伤的细胞模型建立 为建立化学性缺氧损伤的海马细胞模型,本研究首先应用不同浓度的CoCl2对海马细胞作用不同的时间,根据细胞存活率结果确定CoCl2的合适作用浓度和时间。Fig 1A结果显示,300~900 μmol·L-1CoCl2作用海马细胞24 h均可明显降低细胞存活率(P值均<0.05),其中浓度在600 μmol·L-1时,细胞存活率下降至(53.7±2.33)%。随后,我们再观察600 μmol·L-1CoCl2作用海马细胞不同时间对细胞存活率的影响。如Fig 1-B所示,自9 h起,600 μmol·L-1CoCl2呈时间依赖性地降低细胞存活率,在作用36 h时,细胞存活率下降至(52.0±2.65)%,细胞损伤接近半数。因此,本研究确定化学性缺氧损伤海马神经元细胞模型的实验参数为600 μmol·L-1CoCl2作用海马细胞36 h。
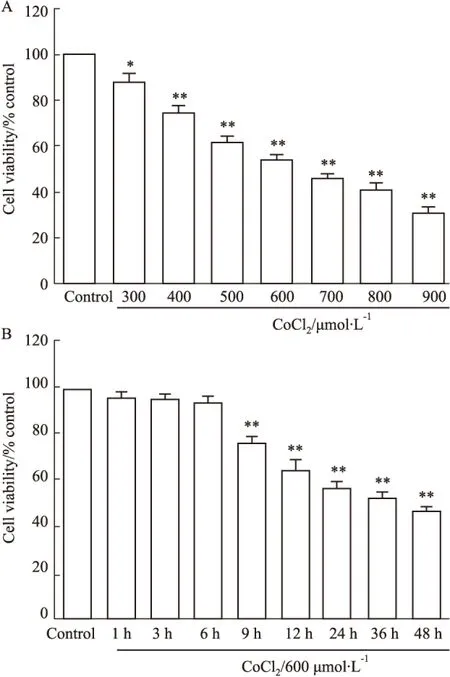
Fig 1 Cobalt chloride (CoCl2) induces a decrease in cell viability in HT22 hippocampal cells (n=3)
HT22 hippocampal cells were treated with CoCl2at different concentrations and different time points.*P<0.05,**P<0.01vscontrol group
2.2 坏死性凋亡介导CoCl2引起的海马细胞存活率降低 如前所述,CoCl2可明显降低使海马细胞存活率。40~100 μmol·L-1Nec-1和CoCl2共处理海马细胞可明显升高细胞存活率,与CoCl2组相比,差异均具有统计学意义(P值均<0.01),其中80 μmol·L-1Nec-1对抗CoCl2降低细胞存活率的作用最明显;不同浓度的Nec-1本身对海马细胞存活率无明显影响(Fig 2)。因此,在后续实验中Nec-1的作用浓度为80 μmol·L-1。
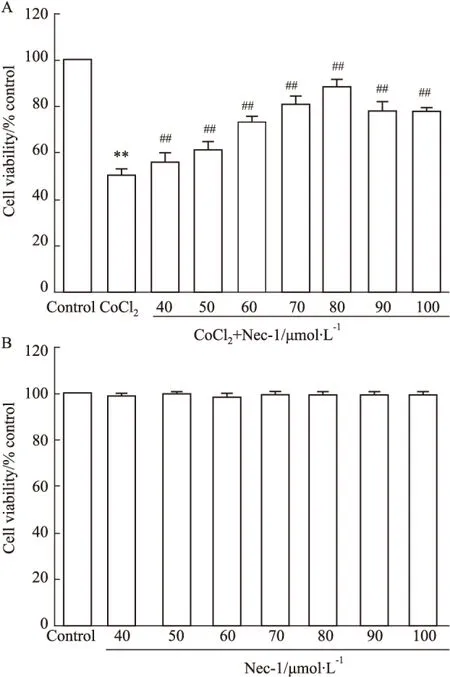
Fig 2 Nec-1 attenuates CoCl2-induced decrease in cell viability in HT22 hippocampal cells (n=3)
A: HT22 hippocampal cells were treated with 600 μmol·L-1CoCl2with or without the co-treatment with Nec-1 (a specific inhibitor of necroptosis) at different concentrations; B:HT22 hippocampal cells were treated with Nec-1 at different concentrations.**P<0.01vscontrol group;##P<0.01vsCoCl2-treated group
2.3 坏死性凋亡介导CoCl2引起的LDH活性增加 Fig 3所示,CoCl2作用海马细胞36 h可使培养液中的LDH活性明显增加,与Control组比较,差异具有统计学意义(P<0.01)。80 μmol·L-1Nec-1和CoCl2共处理海马细胞36 h,LDH活性明显减少,与CoCl2组比较,差异具有统计学意义(P<0.01)。80 μmol·L-1Nec-1本身对LDH的活性无明显的影响。
2.4 坏死性凋亡介导CoCl2引起的海马细胞ROS过度生成 DCFH-DA进入细胞后水解生成的DCFH可被胞内的ROS氧化为发出绿色荧光的DCF,荧光强度可反映细胞ROS的含量。如Fig 4B显示,CoCl2作用海马细胞36 h可使胞内DCF的MFI增加至(30.7±1.27)%,与Control组[Fig 4A,MFI值为(9.63±0.89)%]相比,差异具有统计学意义(P<0.01)。然而,80 μmol·L-1Nec-1和CoCl2共处理海马细胞36 h,可使MFI降低至(13.7±1.41)%(Fig 4C),与CoCl2组比较,差异具有统计学意义(P<0.01)。Nec-1本身对海马细胞ROS的基础生成无明显的影响(P>0.05)。

Fig 3 Nec-1 attenuates CoCl2-induced increase in activity of lactate dehydrogenase (LDH)in HT22 hippocampal cells (n=3)
A: Control; B: CoCl2600 μmol·L-1; C: Nec-1 80 μmol·L-1+ CoCl2600 μmol·L-1; D: Nec-1 80 μmol·L-1.**P<0.01vscontrol group;##P<0.01vsCoCl2-treated group.
Fig 4 Nec-1 attenuates CoCl2-induced increase in intracellular ROS generation in HT22 hippocampal cells (n=3)
A: Control; B: CoCl2600 μmol·L-1; C: Nec-1 80 μmol·L-1+ CoCl2600 μmol·L-1; D: Nec-1 80 μmol·L-1.**P<0.01vscontrol group;##P<0.01vsCoCl2-treated group.
2.5 坏死性凋亡介导CoCl2引起的海马细胞MMP丢失 CoCl2作用海马细胞36 h,可使海马细胞内Rh 123 的MFI从(31.0±1.01)%(Control组,Fig 5A)降低至(10.8±0.92)%(CoCl2组,Fig 5B),两者比较差异有统计学意义(P<0.01)。80 μmol·L-1Nec-1和CoCl2共处理海马细胞36 h,可使MFI升高至(22.0±1.09)%(Fig 5C),与CoCl2组比较,差异有统计学意义(P<0.01)。Nec-1本身对海马细胞MMP无明显的影响(P>0.05)。
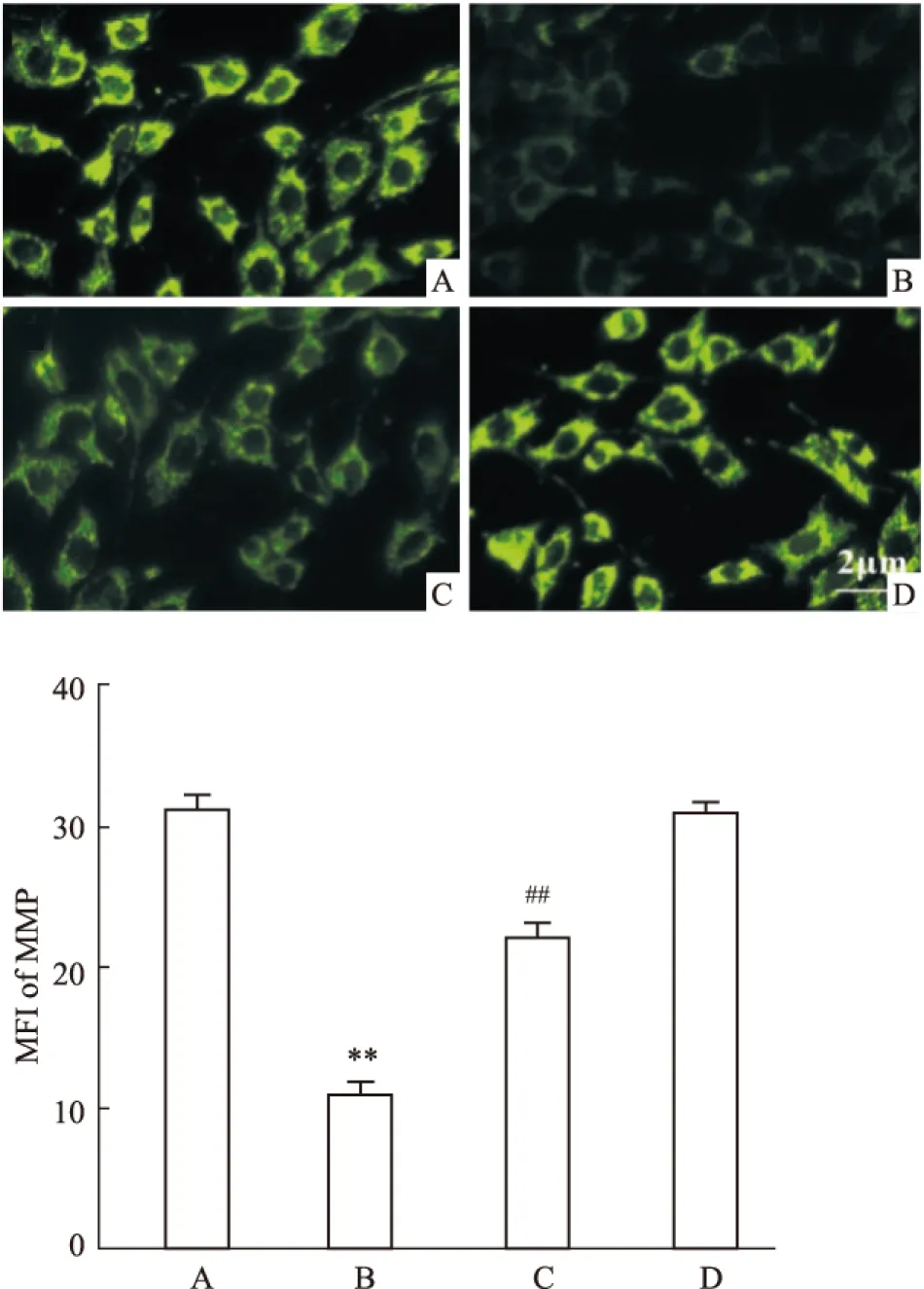
Fig 5 Nec-1 attenuates CoCl2-induced dissipation of mitochondrial membrane potential (MMP) in HT22 hippocampal cells (n=3)
A: Control; B: CoCl2600 μmol·L-1; C: Nec-1 80 μmol·L-1+ CoCl2600 μmol·L-1; D: Nec-1 80 μmol·L-1.**P<0.01vscontrol group;##P<0.01vsCoCl2-treated group.
2.6 坏死性凋亡介导CoCl2引起的海马细胞炎症因子分泌增多 海马细胞经CoCl2作用36 h后,细胞培养液中的炎症因子IL-1β(Fig 6A)和TNF-α(Fig 6B)的分泌明显增多,与Control组比较,差异具有统计学意义(P均<0.01)。然而,80 μmol·L-1Nec-1和CoCl2共处理海马细胞36 h可使IL-1β和TNF-α的分泌明显减少,与CoCl2组比较,差异均具有统计学意义(P均<0.01)。Nec-1本身不影响炎症因子的基础分泌。
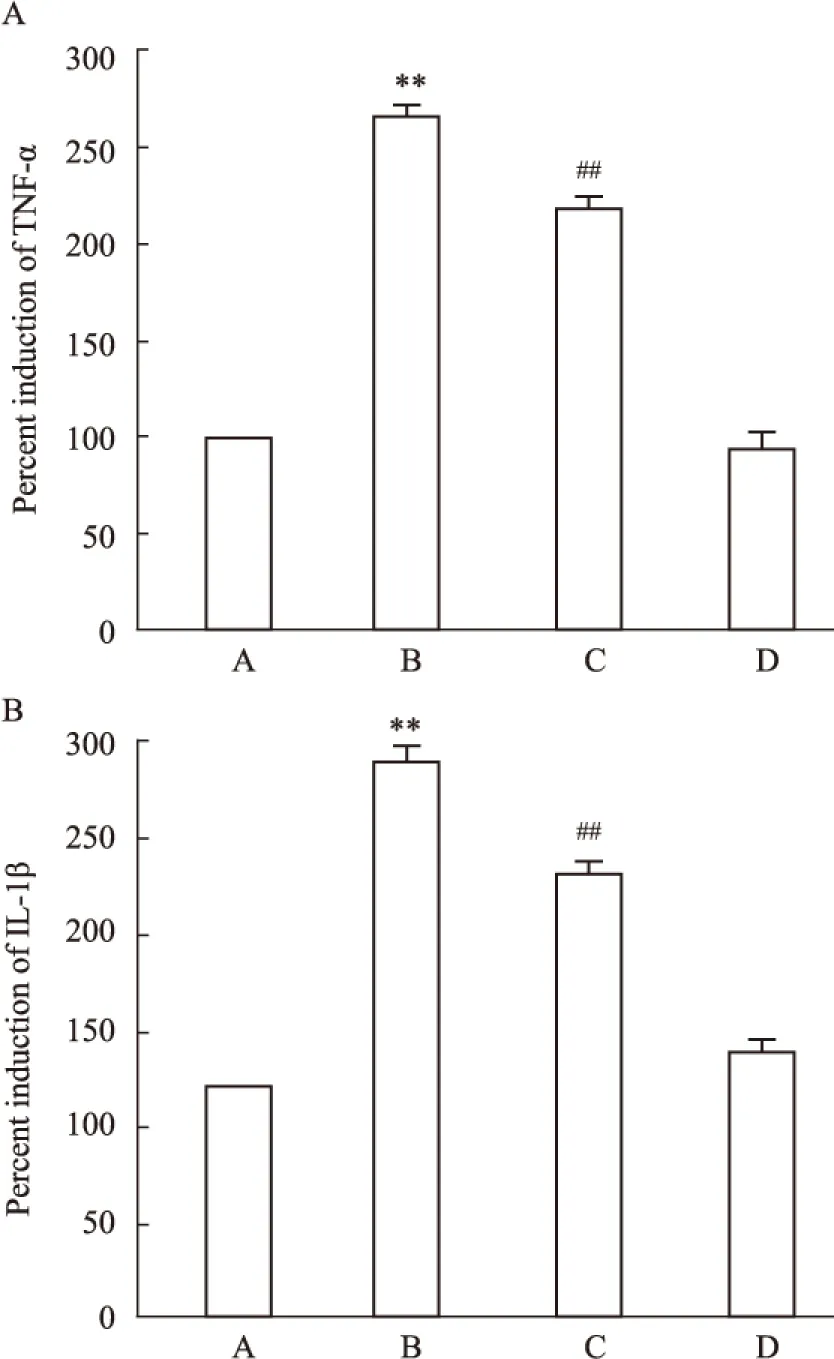
Fig 6 Nec-1 attenuates CoCl2-induced secretion of inflammatory cytokines in HT22 hippocampal cells (n=3)
A: Control; B: CoCl2600 μmol·L-1; C: Nec-1 80 μmol·L-1+CoCl2600 μmol·L-1; D: Nec-1 80 μmol·L-1.**P<0.01vscontrol group;##P<0.01vsCoCl2-treated group.
2.7 Nec-1抑制CoCl2引起的海马细胞RIP3蛋白表达增多 如Fig 7A所示,CoCl2作用海马细胞48 h,其中6 h起RIP3的表达明显增多(P<0.01),随着作用时间延长,RIP3表达进一步增多,12 h时RIP3的表达增多最明显,尽管在24、36和48 h RIP3的表达有所减少,但和Control组比较,差异仍具有统计学意义(P均<0.01)。然而,80 μmol·L-1Nec-1和CoCl2共处理海马细胞12 h,RIP3的表达明显减少,与CoCl2组比较,差异具有统计学意义(P <0.01)。Nec-1本身对RIP3的基础表达无明显的影响(Fig 7B)。
3 讨论
海马结构是与学习和记忆密切联系的重要脑区,根据既往的研究报道,在缺血缺氧损伤时,大脑各分区中以海马区和皮质神经元对缺血缺氧的敏感性尤为明显。因此,本文应用小鼠HT22海马神经元建立化学性缺氧损伤模型探讨海马神经元缺氧的细胞损伤机制。CoCl2是一种化学性缺氧的模拟剂,作用于离体细胞能诱导出缺血/缺氧状态下表现的各种损伤,包括ROS生成增多[4,15-16]、MMP水平下降[4,15-17]及炎症因子分泌增加[18]等。在本研究中,根据细胞存活率结果我们确定应用600 μmol·L-1CoCl2作用HT22海马细胞36 h建立化学性缺氧的神经元损伤模型。与既往研究的结果[4,15-18]相一致,本研究观察到,CoCl2可诱导海马细胞多种损伤,表现为致细胞毒性作用(细胞存活率降低和培养液中LDH活性升高)、氧化应激(ROS生成过多)、线粒体损伤(MMP丢失)及炎症反应(IL-1β和TNF-α的分泌水平增加)。此外,本研究还观察到CoCl2作用海马细胞不同时间均明显地促进RIP3的表达,应用坏死性凋亡的特异性抑制剂Nec-1可抑制CoCl2对RIP3表达的上调,提示化学性缺氧诱导的海马神经元细胞损伤模型中存在坏死性凋亡。坏死性凋亡是在2005年被发现的一种细胞死亡方式,广泛参与心脑血管系统的缺血缺氧性损伤、神经退行性病变等疾病的发生[10-11]。有研究指出[13],小鼠MCAO模型有坏死性凋亡的发生;成年大鼠缺氧损伤后,海马区神经元RIP3表达增多,Nec-1可减轻缺血引起的RIP3表达上调[14],这些结果和本研究的结果是相一致的。
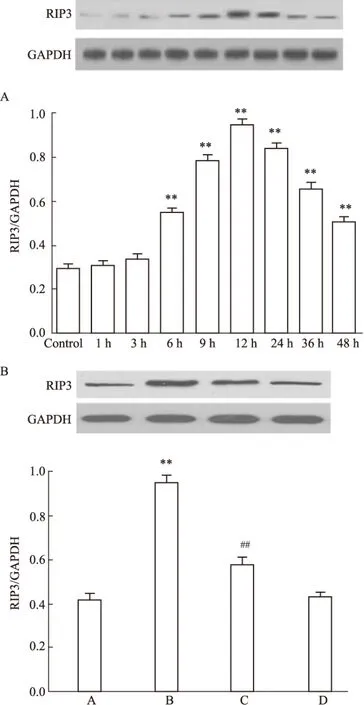
Fig 7 Nec-1 attenuates CoCl2-induced up-regulation of expression of RIP3 in HT22 hippocampal cells (n=3)
A: HT22 hippocampal cells were treated with 600 μmol·L-1CoCl2for a 48 h period; B: HT22 hippocampal cells were treated with 600 μmol·L-1CoCl2for 24 h with or without the co-treatment with 80 μmol·L-1Nec-1. A: Control; B: CoCl2600 μmol·L-1; C: Nec-1 80 μmol·L-1+CoCl2600 μmol·L-1; D: Nec-1 80 μmol·L-1.**P<0.01vscontrol group;##P<0.01vsCoCl2-treated group.
重要的是,各类应激引起的细胞坏死性凋亡与其他的损伤机制包括氧化应激、线粒体损伤、炎症反应等有密切的关系,共同参与细胞的损伤。例如,梁伟杰等[19]最近报道,在高糖(35 mmol·L-1葡萄糖)引起心肌细胞损伤过程中,ROS和坏死性凋亡存在正相互作用,两者相互促进,共同介导心肌细胞的损伤;Lim等指出,Nec-1通过改善线粒体通透性转换孔的功能减少大脑神经元在缺血/再灌注时的缺失[20],表明坏死性凋亡可能对线粒体功能有损伤作用;Liu等[21]研究指出,坏死性凋亡介导创伤性脑损伤引起的小鼠脑部炎症。然而,坏死性凋亡在化学性缺氧诱导的海马神经细胞损伤和炎症中的作用迄今未明。为此,我们观察坏死性凋亡的特异性抑制剂Nec-1对化学性缺氧引起的海马细胞损伤和炎症的影响。研究结果表明,Nec-1可抑制CoCl2引起的多种损害,使细胞存活率升高,LDH活性、ROS生成、MMP丢失及炎症因子的分泌均减少。这些结果提示,坏死性凋亡介导化学性缺氧引起的海马细胞毒性、氧化应激、线粒体损伤和炎症反应。有趣的是,坏死性凋亡的发生通常由TNF-α和相应受体结合而启动,继而激活RIP1、RIP3等一系列信号分子从而引起坏死性凋亡发生;而在本研究中,我们证实坏死性凋亡可促进TNF-α的生成,提示TNF-α和坏死性凋亡之间可能存在着正相互作用,然而,要证实这一推断,尚需进一步的实验论证。
综上所述,本研究在CoCl2诱导HT22海马细胞化学性缺氧的损伤模型中首次证实,坏死性凋亡介导化学性缺氧引起的海马细胞损伤和炎症反应,这可能是化学性缺氧引起神经损害的一个重要机制。
(致谢:本实验在南华大学生理教研室与南华大学医学院病理实验室开展,衷心感谢梁伟杰老师在实验中给予的大力支持!)
[1] Meng J L, Mei W Y, Dong Y F, et al. Heat shock protein 90 mediates cytoprotection by H?S against chemical hypoxia-induced injury in PC12 cells[J].ClinExpPharmacolPhysiol, 2011, 38(1): 42-9.
[2] Lan A, Liao X, Mo L, et al. Hydrogen sulfide protects against chemical hypoxia-induced injury by inhibiting ROS-activated ERK1/2 and p38MAPK signaling pathways in PC12 cells[J].PLoSOne, 2011, 6(10): e25921.
[3] Wei R, Zhang R, Xie Y, et al. Hydrogen suppresses hypoxia/reoxygenation-induced cell death in hippocampal neurons through reducing oxidative stress[J].CellPhysiolBiochem, 2015, 36(2): 585-598.
[4] 徐文明,兰爱平,林建聪,等. 活性氧激活的ERK1/2通路在化学性缺氧损伤PC12细胞中的作用[J]. 中国药理学通报, 2013, 29(7): 985-90.
[4] Xu W M, Lan A P, Lin J C, et al. Role of ROS-activated ERK1/2 pathway in chemical hypoxia-induced injury in PC12 cells[J].ChinPharmacolBull, 2013, 29(7): 985-90.
[5] Chhunchha B, Fatma N, Kubo E, et al. Curcumin abates hypoxia-induced oxidative stress based-ER stress-mediated cell death in mouse hippocampal cells (HT22) by controlling Prdx6 and NF-κB regulation[J].AmJPhysiolCellPhysiol, 2013, 304(7): C636-C655.
[6] Wang W M, Liu Z, Liu A J, et al. The zinc ion chelating agent TPEN attenuates neuronal death/apoptosis caused by hypoxia/ischemia via mediating the pathophysiological cascade including excitotoxicity, oxidative stress, and inflammation[J].CNSNeurosciTher, 2015, 21(9): 708-17.
[7] Gong HY, Zheng F, Zhang C, et al. Propofol protects hippocampal neurons from apoptosis in ischemic brain injury by increasing GLT-1 expression and inhibiting the activation of NMDAR via the JNK/Akt signaling pathway[J].IntJMolMed, 2016, 38(3): 943-50.
[8] Wu L Y, Ma Z M, Fan X L, et al. The anti-necrosis role of hypoxic preconditioning after acute anoxia is mediated by aldose reductase and sorbitol pathway in PC12 cells[J].CellStressChaperones, 2010, 15(4): 387-394.
[9] Descloux C, Ginet V, Clarke P G, et al. Neuronal death after perinatal cerebral hypoxia-ischemia: Focus on autophagy-mediated cell death[J].IntJDevNeurosci, 2015, 45: 75-85.
[10] Degterev A, Huang Z, Boyce M, et al. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury[J].NatChemBiol, 2005, 1(2): 112-9.
[11] Degterev A, Zhou W, Maki J L, et al. Assays for necroptosis and activity of RIP kinases[J].MethodsEnzymol, 2014, 545: 1-33.
[12] Cho Y S, Challa S, Moquin D, et al. Phosphorylation-driven assembly of the RIP1-RIP3 complex regulates programmed necrosis and virus-induced inflammation[J].Cell, 2009, 137(6): 1112-23.
[13] Kelliher M, Grimm S, Ishida Y, et al. The death domain kinase RIP mediates the TNF-induced NF-kappaB signal[J].Immunity, 1998, 8(3): 297-303.
[14] Vieira M, Fernandes J, Carreto L, et al. Ischemic insults induce necroptotic cell death in hippocampal neurons through the up-regulation of endogenous RIP3[J].NeurobiolDis, 2014, 68: 26-36.
[15] Zou W, Yan M, Xu W, et al. Cobalt chloride induces PC12 cells apoptosis through reactive oxygen species and accompanied by AP-1 activation[J].JNeurosciRes, 2001, 64(6): 646-53.
[16] Ha J S, Park S S. Glutamate-induced oxidative stress, but not cell death, is largely dependent upon extracellular calcium in mouse neuronal HT22 cells[J].NeurosciLett, 2006, 393(2-3): 165-9.
[17] 华潇潇,陈洁琳,张莉莉,等. 右美托咪定抑制化学性低氧引起的PC12细胞炎症反应[J]. 中国药理学通报, 2014, 30(1): 85-9.
[17] Hua X X, Chen J L, Zhang L L, et al. Dexmedetomidine inhibits inflammation induced by chemical hypoxia in PC12 cells[J].ChinPharmacolBull, 2014, 30(1): 85-9.
[18] Lan A, Xu W, Zhang H, et al. Inhibition of ROS-activated p38MAPK pathway is involved in the protective effect of H2S against chemical hypoxia-induced inflammation in PC12 cells[J].NeurochemRes, 2013, 38(7): 1454-66.
[19] 梁伟杰,何洁仪,陈景福,等. 坏死性凋亡与活性氧的相互作用介导高糖引起的H9c2心肌细胞损伤[J]. 中国动脉硬化杂志, 2016, 24(8): 781-7.
[19] Liang W J, He J Y, Chen J F, et al. The interaction between necroptosis and reactive oxygen species mediates high glucose-induced injury in H9c2 cardiac cells[J].ChinJArterioscler, 2016, 24(8): 781-7.
[20] Lim S Y, Davidson S M, Mocanu M M, et al. The cardioprotective effect of necrostatin requires the cyclophilin-D component of the mitochondrial permeability transition pore[J].CardiovascDrugsTher, 2007, 21(6): 467-9.
[21] Liu T, Zhao D X, Cui H, et al. Therapeutic hypothermia attenuates tissue damage and cytokine expression after traumatic brain injury by inhibiting necroptosis in the rat[J].SciRep, 2016, 6: 24547.
Necroptosis mediates chemical hypoxia-induced injury and inflammation in HT22 hippocampal cells
WANG Bo1, XU Yong1, LI Xiang1, HOU Jiao-yan1, ZHOU Zhong-qun2, TIAN Shao-wen3, KUANG Xin1
(1.DeptofAnesthesiology; 2.DeptofPain,FirstAffiliatedHospital,UniversityofSouthChina,HunanHengyang421001,China; 3.DeptofPhysiology,MedicalSchool,UniversityofSouthChina,HunanHengyang421001,China)
Aim To investigate whether necroptosis mediates chemical hypoxia-induced HT22 mouse hippocampal cell injury and inflammation. Methods HT22 hippocampal cells were exposed to cobalt chloride (CoCl2) to establish a model of the chemical hypoxia-induced injury and inflammation. The expression level of RIP3 (an index of necroptosis) was determined by Western blot. Cell counter kit-8 (CCK-8) assay was used to test the cell viability. Lactate dehydrogenase (LDH) activity in the culture medium was measured with commercial kits. Mitochondrial membrane potential (MMP) was examined by rhodamine123 staining followed by photofluorography. The intracellular level of reactive oxygen species (ROS) was detected by 2’, 7’-dichlorfluorescein-diacetate (DCFH-DA) staining followed by photofluorography. The secretion levels of interleukin-1β (IL-1β) and tumor necrosis factor-a (TNF-α) were measured by ELISA. Results Treatment of HT22 hippocampal cells with 600 μmol·L-1CoCl2for 36 h markedly induced cytotoxicity, leading to a decrease in cell viability to (52.0±2.65) % , indicating that chemical hypoxia-induced cellular injury model was successfully set up. Besides, CoCl2induced considerable injuries and inflammation, evidenced by increases in LDH activity, ROS production, MMP loss, as well as the secretion levels of IL-1β and TNF-α. Co-treatment of the cells with 40~100 μmol·L-1Nec-1 (a specific inhibitor of necroptosis) and CoCl2markedly attenuated the decrease in viability induced by CoCl2, reaching the best anti-cytotoxicity inhibitory effect at 80 μmol·L-1. Meanwhile, the co-treatment with 80 μmol·L-1Nec-1 blocked the above injuries and inflammatory response induced by CoCl2. In addition, treatment of HT22 hippocampal cells for 6~48 h up-regulated the expression of RIP3, and Nec-1 alleviated the up-regulation of RIP3 expression level induced by CoCl2. Conclusion Necroptosis mediates chemical hypoxia-induced HT22 hippocampal cell injury and inflammation.
necroptosis; chemical hypoxia; cobalt chloride; HT22 hippocampal cell; injury; inflammation
时间:2017-3-13 8:38
http://kns.cnki.net/kcms/detail/34.1086.R.20170324.1247.016.html
2016-11-30,
2017-02-03
国家自然科学基金资助项目(No 8140050613);湖南省教育厅资助课题(No 15C1208)
王 波(1987-),男,硕士,住院医师,研究方向:脑保护与认知功能,Tel:0734-8578506,E-mail:277433307@qq.com; 徐 勇(1991-),女,硕士生,研究方向:脑保护与认知功能,并列第一作者,E-mail:478668259@qq.com; 旷 昕(1971-),男,博士,主任医师、硕士生导师,研究方向:认知功能障碍与脑保护,神经病理性疼痛,通讯作者,Tel:0734-8578506,E-mail:kx6924@126.com
10.3969/j.issn.1001-1978.2017.04.008
A
1001-1978(2017)04-0480-07
R-332;R322.81;R329.24;R364.5;R392.12;R845.22
