含氮杂环配合物的光学性质理论研究
2017-04-12赵威
赵 威
(营口理工学院化学与材料工程系,辽宁营口 115004)
含氮杂环配合物的光学性质理论研究
赵 威
(营口理工学院化学与材料工程系,辽宁营口 115004)
在密度泛函理论(DFT)的B3LYP/6-31G(d,p)水平上,对含氮杂环配合物以1,3,5-三嗪环为单体,分别加入卤素Cl,噻吩环或者对称的加入噻吩环后四种含氮杂环化合物的几何结构进行了全优化。讨论缺电子类含氮杂环配合物进行修饰后的结构,对在获得基态稳定构型的基础上,应用含时密度泛函理论(TD-DFT)计算设计化合物的电子吸收光谱,讨论了最大吸收波长和部分前线分子轨道能量,以期为同类化合物的合成提供理论依据。
密度泛函理论;稳定构型;吸收光谱
引言
近年来高分子共轭化合物的光电研究越来越受到研究者们的关注,尤其对一些含缺电子杂环类化合物的研究成为热点。关于富电子环和缺电子环构成的聚合物具有电荷移动的性质,S0和S1之间的能量有所降低,可以合成一些电荷移动型聚合物。1,3,5-三嗪环上可以进行化学修饰,设计一些列新的化合物,比如:加入卤素Cl,噻吩环或者对称的加入噻吩环,对其设计的化合物进行光谱性质研究,对于开发这一类化合物提供理论指导作用。计算结构见图1。
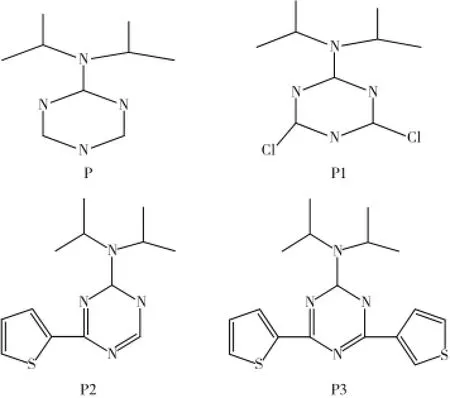
图1 含氮杂环配合物
1 计算结果与讨论
所有的计算均采用高斯Gaussian03计算程序完成,首先对四种化合物进行了基态几何结构优化,进行频率计算来确认设计的化合物稳定存在。
1.1 光谱计算
应用含时密度泛函理论(TD-DFT)计算设计化合物的电子吸收光谱,分别计算出了含氮杂环化合物的跃迁轨道、成分、最大吸收波长(λ)、振子强度(f)和主要跃迁类型见表1。
计算结果表明四种含氮杂环化合物的跃迁轨道均为HOMO到LUMO轨道,对应的跃迁为基态S0到激发态S1的π-π*跃迁,其中化合物P3的最大吸收波长最长为333.94 nm,其次是含氮杂环化合物P2的最大吸收波长为314.06nm,化合物P1和化合物P的最大吸收波长几乎相近分别为270.23nm和273.43nm,四种含氮杂环化合物均属于紫外光波长范围。

表1 含氮杂环化合物的跃迁轨道、成分、λ、f和主要跃迁类型
对于发光材料的配物来说,对其进行前线分子轨道比较重要。结合前线分子轨道数据可以分析化合物的发光机理,进一步解释最大吸收波长移动的原理,四种含氮杂环化合物的部分前线分子轨道能量数据见表2。
2 结论
分子的前线轨道分为HOMO和LUMO轨道,HOMO轨道表示已经占有电子的能级最高的轨道,也称为最高占有轨道,LUMO轨道表示未占有电子的能级最低的轨道。HOMO与LUMO之间的能量差称为“能带隙”,这个能量差即称为HOMO-LUMO的跃迁能级,化合物的跃迁能级可以用来衡量一个分子是否容易被激发:LUMO与HOMO之间的带隙越小,表明分子越容易被激发。四种化合物中P1化合物的HOMO轨道能量最高,LUMO轨道能量比较低,电子不容易跃迁,所以P1化合物能隙比较大。P2化合物的HOMO轨道能量最低,LUMO轨道能量比较高,电子容易跃迁,所以P2化合物能隙比较小仅为0.168 13e V。
[1] 刘方明,鲁文杰,张正方,等.含2一三氟甲基苯并咪哇杂环化合
物的合成[J].高等学校化学学报,1999,20(8):1242-1247.
[2] 金春华,罗荣.5-甲氧基-2-疏基苯并眯哇合成新工艺[J].精细化工,1999,16(3):52.
Theoretical study on optical properties of nitrogen containing heterocyclic complexes
Zhao Wei
Based on the density functional theory(DFT)B3LYP/6-31G(D,P)on the level of nitrogen heterocyclic complexes with 1,3,5-three Thiotriazinone as monomers,respectively adding halogen Cl,thiophene ring geometry or symmetric join thiophene ring after four kinds of nitrogenous heterocyclic compounds were optimized and discussed the electron deficient nitrogenous heterocyclic complexes structure was modified,based on the ground state of the stable structure,using the time-dependent density functional theory(TD-DFT)calculation of the electronic absorption spectra of compounds is discussed,the maximum absorption wavelength and frontier molecular orbital energy,in order to provide a theoretical basis for the synthesis of similar compounds.
DFT;Stable configuration;absorption wavelength

表2 四种含氮杂环化合物的部分前线分子轨道能量
UDC
A
1003-6490(2017)01-0097-02
2016-11-16
赵威(1984—),女,辽宁盖州人,硕士研究生,主要研究方向物理化学大分子功能材料。
