液相色谱-串联质谱法测定大鼠血浆中靛蓝的浓度及药动学研究
2017-04-12孔树佳李砚文昆明医科大学第三附属医院云南省肿瘤医院药剂科昆明650118
孔树佳,李砚文(昆明医科大学第三附属医院/云南省肿瘤医院药剂科,昆明 650118)
液相色谱-串联质谱法测定大鼠血浆中靛蓝的浓度及药动学研究
孔树佳*,李砚文#(昆明医科大学第三附属医院/云南省肿瘤医院药剂科,昆明 650118)
目的:建立测定大鼠血浆中靛蓝浓度的方法,研究靛蓝在大鼠体内的药动学特征。方法:将18只大鼠随机分为低、中、高剂量组,每组6只,分别ig 10、20、40 mg/kg的靛蓝溶液。分别于给药前和给药后0.083、0.25、0.5、0.75、1、2、4、6、8、10、12、16、24、48、72 h眼眶采全血0.3 mL,分离血浆,甲醇沉淀后采用液相色谱-串联质谱(LC-MS/MS)法测定靛蓝的血药浓度。色谱柱为Agilent Poroshell EC-C18,流动相为甲醇-5 mmol/L乙酸铵溶液(95∶5,V/V),流速为0.4 mL/min;以多反应监测(MRM)模式进行定量分析,用于监测的离子对(m/z)为263.1~218.8(靛蓝)和237.2~194.1(卡马西平,内标);采用DAS 3.0软件计算药动学参数。结果:靛蓝检测质量浓度的线性范围为0.5~100 ng/mL(r=0.999 9),日内、日间RSD均小于9%(n=5),低、中、高质量浓度的质控样品的基质效应分别为(98.25±3.71)%、(102.23±2.64)%、(102.29±3.79)%(n=5)。靛蓝在低、中、高剂量组大鼠体内的tmax分别为(8.6±1.1)、(9.2±0.8)、(9.5±0.8)h,cmax分别为(30.9±8.6)、(44.9±10.1)、(96.1±17.4)ng/mL,t1/2分别为(14.9±2.1)、(16.3± 2.9)、(15.3±3.7)h,AUC0-72h分别为(366.6±83.4)、(694.9±105.8)、(1 223.42±108.7)ng·h/mL。结论:本方法灵敏度高、特异性好,可用于大鼠血浆样品中靛蓝的含量测定;靛蓝在大鼠体内药动学特征符合非房室模型。
靛蓝;液相色谱-串联质谱法;大鼠;血浆;药动学
青黛,别名靛花、蓝靛,主要含靛蓝、靛玉红、色胺酮和青黛酮等活性成分,具有清热解毒、消肝泻火、凉血止血、定惊之功效。近年来,青黛类中药被广泛用于内科、外科、儿科、妇科、传染科等病症的治疗,引起了国内外研究者的重视[1]。靛蓝是青黛的主要成分之一,具有抗病毒作用[2],对幽门螺旋杆菌的体外抗菌活性较好[3-4],但其体内过程文献报道较少。目前对于靛蓝的分析方法主要有高效液相色谱(HPLC)法[5]、气相色谱-质谱联用(GC-MS)法[6]和液相色谱-质谱联用(LC-MS)法[7],大多用于中药质量标准评价或体外试验测定,样品分析时间长、样品前处理方法复杂、定量下限高,均不太适合用于生物样本分析及体内过程评价。在本研究中,笔者拟以卡马西平为内标,建立以液相色谱-电喷雾串联质谱(LC-MS/MS)法[8]测定大鼠血浆中靛蓝含量的分析方法,并将其用于靛蓝在大鼠体内的药动学研究。
1 材料
1.1 仪器
5500 Q TRAPTM质谱仪,配有电喷雾(ESI)离子源(美国AB Sciex公司);30A系列超HPLC仪(日本岛津公司);H1650-W高速台式离心机(湖南湘仪实验室开发有限公司,离心半径:20 cm)。
1.2 药品与试剂
靛蓝对照品(上海源叶生物科技有限公司,批号:110716-201111,纯度:≥98%);卡马西平对照品(中国食品药品检定研究院,批号:100142-201004,纯度:≥99%);甲醇、乙酸铵为色谱纯,其他试剂均为分析纯,水为Milli-Q超纯水。
1.3 动物
SD大鼠,♂,体质量200~220 g,购自第三军医大学实验动物中心,动物合格证号为SCXK(军)2007-015、SCXK(渝)2007-0003。实验所使用大鼠的购买、实验处置都严格遵循《第三军医大学实验动物管理暂行规定》。
2 方法与结果
2.1 溶液的制备
2.1.1 对照品溶液 精密称取靛蓝对照品适量(相当于靛蓝10.0 mg),置于10 mL量瓶中,用甲醇溶解并稀释制成质量浓度为1.0 mg/mL的贮备液。精密量取上述贮备液10µL,加入990µL甲醇,涡旋混匀,得10 μg/mL的对照品溶液,-20℃下保存,备用。
2.1.2 内标溶液 精密称取卡马西平对照品10.11 mg(相当于卡马西平10.0 mg),置于10 mL量瓶中,用甲醇溶解并稀释制成质量浓度为1.0 mg/mL的贮备液。精密量取上述贮备液10µL,加入990µL甲醇,涡旋混匀,制得10 μg/mL的内标溶液,-20℃下保存,备用。
2.2 色谱条件与质谱条件
色谱柱:Agilent Poroshell EC-C18(150 mm×3.0 mm,2.7 μm);流动相:甲醇-5 mmol/mL乙酸铵溶液(95∶5,V/V,pH 6.8),流速:0.4 mL/min;柱温:25℃;进样量:2 μL。
靛蓝和卡马西平的离子化采用ESI离子源,正离子模式,以多反应监测(MRM)模式分析。离子源参数:气帘气为39 psi(1 psi=6 895 Pa),雾化气为88 psi,辅助加热气为89 psi,离子源温度为500℃,离子源电压为5 000 V。四级杆参数:靛蓝监测离子对(m/z)为263.1~218.8,去簇电压(DP)为60 eV,碰撞能量(CE)为33 eV,碰撞池出口电压(CXP)为3.3 eV;卡马西平监测离子对(m/z)为237.2~194.1,DP为100 eV,CE为26.3 eV,CXP为5.9 eV。
2.3 样品处理
将40 ng/mL内标甲醇溶液1 mL加入到0.1 mL样品中,涡旋混匀2 min,13 000 r/min离心5 min,取上清过0.22 μm微孔滤膜,滤液供LC-MS/MS上样分析。
2.4 方法学考察
2.4.1 专属性试验 按相关专属性试验方法检测发现,在正离子模式下,靛蓝和内标产生了质子化的母离子[M+1]+。质谱图中可观察到,靛蓝主要的离子为m/z 263.1,内标主要的离子为m/z 237.2;靛蓝强度最高的子离子为m/z 218.8,内标强度最高的子离子为m/z 194.1。在“2.2”项条件下,内源性杂质对待测物无干扰,靛蓝和内标峰形良好,保留时间分别为2.92、2.16 min。靛蓝和内标的质谱图见图1,LC-MS/MS图见图2。 40、20、5、2、1、0.5 ng/mL的质控样品,按“2.3”项下方法处理后,进样测定。以靛蓝的峰面积和内标峰面积的比值(y)与靛蓝的质量浓度(x)进行回归分析,得回归方程为y=0.049 3x+0.015 9(r=0.999 9)。结果表明,靛蓝检测质量浓度的线性范围为0.5~100 ng/mL,定量下限为0.5 ng/mL。
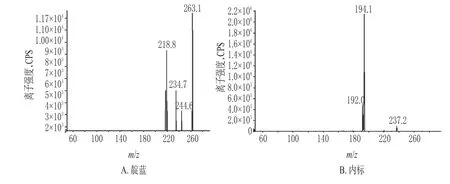
图1 靛蓝和内标的质谱图Fig 1 Spectrometry of indigo and internal standard
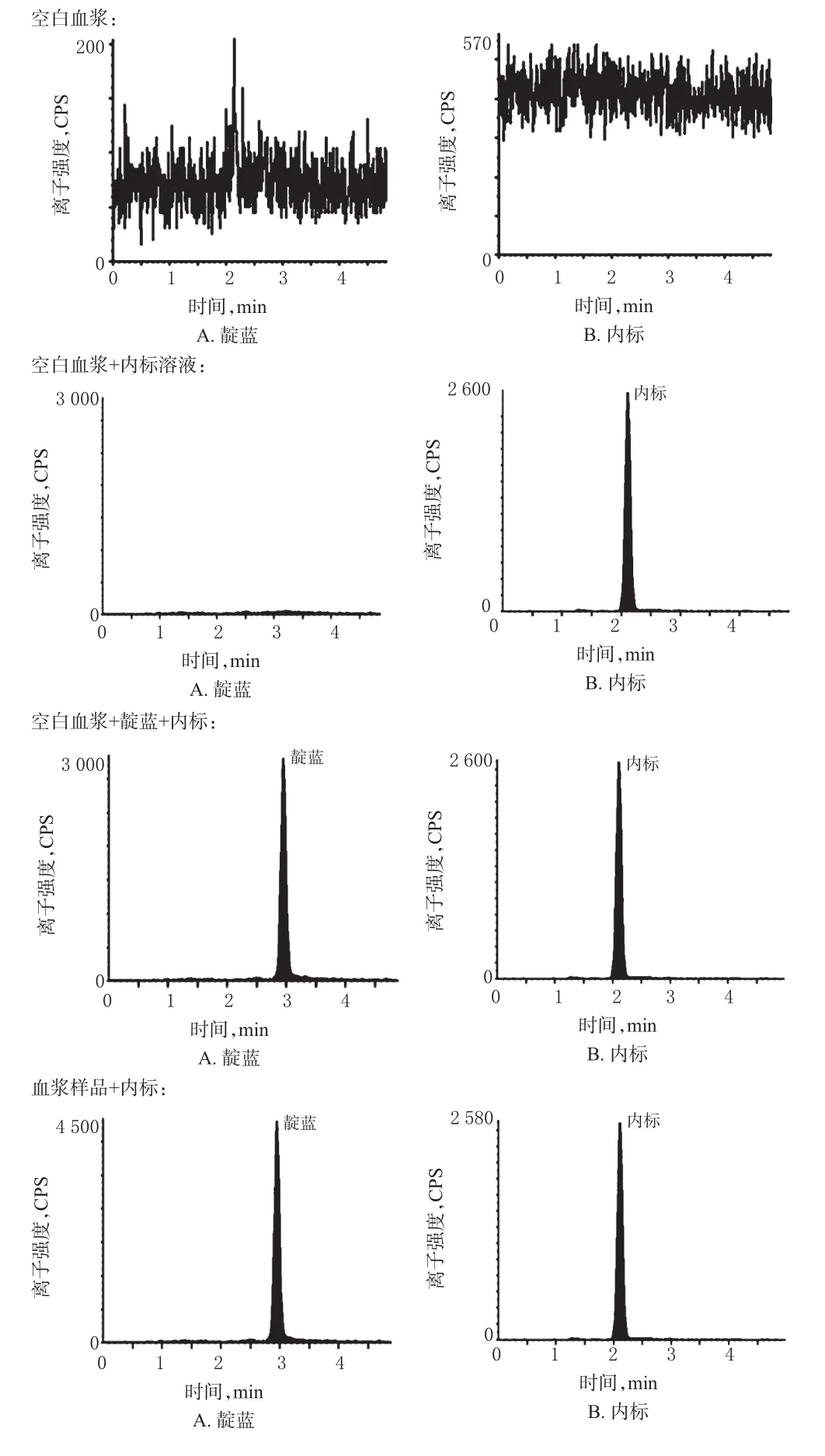
图2 液相色谱-串联质谱图Fig 2 LC-MS/MS spectrometry
2.4.3 基质效应考察 分别以甲醇沉淀后的大鼠空白血浆和去离子水为溶剂,制备靛蓝低、中、高质量浓度(1、10、80 ng/mL)的样品,每个质量浓度5份,按“2.3”项下方法处理后,进样测定。基质效应(%)=沉淀后空白血浆制备样品的峰面积/去离子水制备样品的峰面积× 100%;内标归一化的基质效应(%)=靛蓝的基质效应/内标的基质效应×100%。基质效应考察结果见表1。
表1 基质效应考察结果(±s,n=5)Tab 1 Results of matrix effects(±s,n=5)
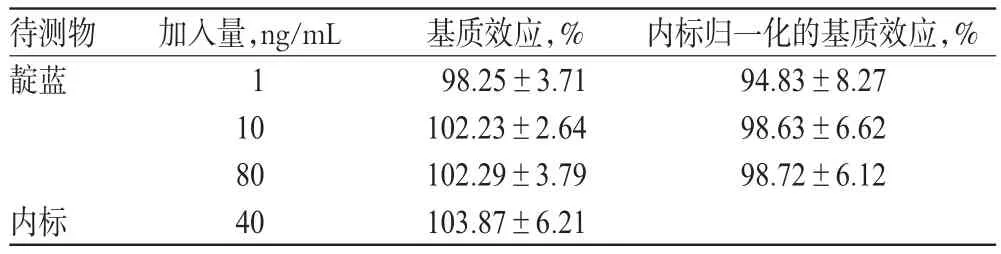
表1 基质效应考察结果(±s,n=5)Tab 1 Results of matrix effects(±s,n=5)
待测物靛蓝内标归一化的基质效应,% 94.83±8.27 98.63±6.62 98.72±6.12加入量,ng/mL 1 10 80 40内标基质效应,% 98.25±3.71 102.23±2.64 102.29±3.79 103.87±6.21
2.4.2 线性关系与定量下限考察 取大鼠空白血浆加入适量的靛蓝对照品溶液,分别制备成含靛蓝100、50、
2.4.4 精密度和准确度试验 制备低、中、高质量浓度(1、10、80 ng/mL)的质控样品,每个质量浓度5份,按“2.3”项下方法处理后,进样测定,连续测定3 d,考察日内、日间精密度和准确度。结果显示,低、中、高质量浓度质控样品的日内RSD分别为8.13%、1.93%、2.48%(n=5),日间RSD分别为3.27%、4.36%、4.85%(n=3),准确度分别为94.2%、103.2%、108.72%(n=5)。
2.4.5 稳定性试验 制备低、中、高质量浓度(1、10、80 ng/mL)的质控样品,分别置于室温下4 h(n=5)、自动进样器内24 h(n=5)、-80℃下14 d(n=5)、-80℃反复冻融3次(n=5),再按“2.3”项下方法处理后进样测定,考察稳定性。结果显示,RSD均小于15%,表明靛蓝的血浆样品在上述条件下均稳定。
2.5 药动学实验
将18只大鼠随机分为低、中、高剂量组,每组6只,分别ig 10、20、40 mg/kg的靛蓝溶液。分别于给药前和给药后0.083、0.25、0.5、0.75、1、2、4、6、8、10、12、16、24、48、72 h眼眶采血0.3 mL并抗凝,常温下4 000 r/min(离心半径:20 cm)离心5 min分离血浆后,-80℃下冻存。采用LC-MS/MS法测定血浆中靛蓝的浓度,绘制药-时曲线。各组大鼠体内靛蓝的药-时曲线见图3。
采用DAS 3.0软件,按非房室模型测定靛蓝的主要药动学参数。结果显示,靛蓝在大鼠体内9 h左右达到峰浓度,t1/2约为15 h,cmax、AUC0-72h、AUC0-∞相对于给药剂量呈近似线性的变化。各组大鼠体内靛蓝的药动学参数见表2。
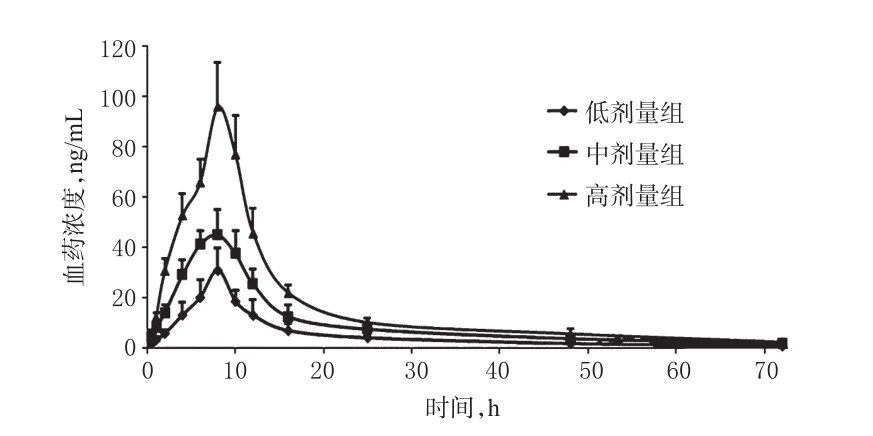
图3 各组大鼠体内靛蓝的药-时曲线(n=6)Fig 3 Concentration-time curves of indigo in rats in vivo in each group(n=6)
表2 各组大鼠体内靛蓝的药动学参数(±s,n=6)Tab 2 Pharmacokinetic parameters of indigo in rats in vivo in each group(±s,n=6)
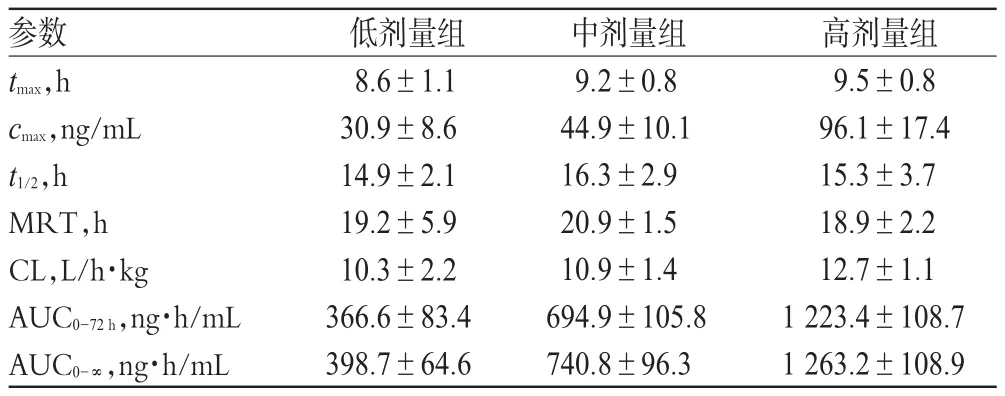
表2 各组大鼠体内靛蓝的药动学参数(±s,n=6)Tab 2 Pharmacokinetic parameters of indigo in rats in vivo in each group(±s,n=6)
参数tmax,h cmax,ng/mL t1/2,h MRT,h CL,L/h·kg AUC0-72h,ng·h/mL AUC0-∞,ng·h/mL低剂量组8.6±1.1 30.9±8.6 14.9±2.1 19.2±5.9 10.3±2.2 366.6±83.4 398.7±64.6中剂量组9.2±0.8 44.9±10.1 16.3±2.9 20.9±1.5 10.9±1.4 694.9±105.8 740.8±96.3高剂量组9.5±0.896.1±17.415.3±3.718.9±2.212.7±1.1 1 223.4±108.7 1 263.2±108.9
3 讨论
3.1 色谱条件优化
大鼠血浆中靛蓝测定方法建立过程中,提高检测的灵敏度是一个比较关键的问题。本实验参考文献[9-10]对方法建立过程的多个环节进行了优化,使得定量下限为0.5 ng/mL。
笔者比较了水、乙腈、甲醇、甲醇-乙腈(1∶1,V/V)、甲醇-水(1∶1,V/V)、乙腈-水(1∶1,V/V)和甲醇-乙腈-水(1∶1∶1,V/V/V)、含0.1%甲酸的甲醇、含0.1%甲酸的乙腈、含0.1%甲酸的水、含0.1%甲酸的甲醇-乙腈(1∶1,V/ V)、含0.1%甲酸的甲醇-水(1∶1,V/V)、含0.1%甲酸的乙腈-水(1∶1,V/V)和含0.1%甲酸的甲醇-乙腈-水(1∶1∶1,V/V/V)对靛蓝对照品的溶解效果,发现甲醇的溶解效果最好,且超声(60 Hz,40℃)就能产生助溶效果。
笔者比较了乙腈和甲醇对待测物和内标的洗脱效果,发现甲醇洗脱所得色谱峰更窄、峰形更好、灵敏度更高;比较了甲醇-水和甲醇-乙酸铵缓冲盐体系,发现若体系中没有乙酸铵,则有拖尾现象,而在流动相中添加5 mmol/L乙酸铵后,对峰形有较大的改善。
笔者比较了等度洗脱[甲醇-5 mmol/L乙酸铵溶液(体积比依次为80∶20、85∶15、90∶10、95∶5)和甲醇]和梯度洗脱的效果,发现甲醇-5 mmol/L乙酸铵溶液(95∶5,V/V)洗脱时灵敏度最高,且出峰时间最短。
3.2 给药剂量的设定
在前期的预实验中,笔者进行了给药剂量的摸索,分别给予5、10、20、40、80 mg/kg,每个剂量组2只大鼠。结果发现5 mg/kg给药剂量所测得的药-时曲线上的点有1/3在本方法的定量下限以下,而80 mg/kg给药剂量所得的药-时曲线在tmax附近的点有超限的现象,在10~40 mg/kg的给药剂量范围内所测定的血药浓度都在工作曲线的线性范围内,故本实验采用10、20、40 mg/kg为给药剂量。
3.3 取血时间点的设定
在前期的预实验中,笔者通过每组2只大鼠所测定的血药浓度,采用DAS 3.0软件处理后获得的tmax在8.5 h左右。根据采样时间点的设计原则,即兼顾药物的吸收相(吸收相至少需要2~3个采样点)、平衡相(在cmax附近至少需要3个采样点)和消除相(需要4~6个采样点),整个采样时间至少应持续到3~5个半衰期,据此笔者设定了本研究的采血时间点。
[1] Stasiak N,Kukula-Koch W,Glowniak K.Modern industrial and pharmacological applications of indigo dye and its derivatives:a review[J].Acta Pol Pharm,2014,71(2):215-221.
[2] 奇锦峰,孙晨,王永辉,等.板蓝根及所含靛蓝和靛玉红强烈抑制小鼠肾主要有机阴离子转运体Oat1,Oat2和Oat3
[J].中国药理学与毒理学杂志,2014,28(6):878-886.
[3] 杜平华,朱世真,吕品.20种中药材对幽门螺杆菌体外抗菌活性的研究[J].中药材,2001,24(03):188-189.
[4] Chiang YR,Li A,Leu YL,et al.An in vitro study of the antimicrobial effects of indigo naturalis prepared from Strobilanthes formosanus Moore[J].Molecules,2013,18(11):14381-14396.
[5] 胡艳红,李志浩.HPLC测定小儿清咽颗粒中靛蓝和靛玉红的含量[J].现代仪器与医疗,2013,19(4):58-61.
[6] Degani L,Riedo C,Chiantore O.Identification of natural indigo in historical textiles by GC-MS[J].Anal Bioanal Chem,2015,407(6):1695-1704.
[7] Tsuji S,Nakano M,Furukawa M,et al.Identification and determination of the isomer and subsidiary color in food blue no.2(indigo carmine)by LC/MS and HPLC[J]. Shokuhin Eiseigaku Zasshi,2005,46(3):116-120.
[8] 梅升辉,罗旭颖,李倩,等.LC-MS/MS法测定人血浆中替加环素的浓度及其临床应用[J].中国药房,2016,27(5):612-615.
[9] Pauk V,Havlicek V,Papouskova B,et al.Simultaneous identification of historical pigments Prussian blue and indigo in paintings by electrospray mass spectrometry[J].J Mass Spectrom,2013,48(8):927-930.
[10] Lu HT,Liu J,Deng R,et al.Preparative isolation and purification of indigo and indirubin from Folium isatidis by high-speed counter-current chromatography[J].Phytochem Anal,2012,23(6):637-641.
(编辑:邹丽娟)
Concentration Determination of Indigo in Rats’Plasma by LC-MS/MS and Its Pharmacokinetics Study
KONG Shujia,LI Yanwen(Dept.of Pharmacy,the Third Affiliated Hospital of Kunming Medical University/ Yunnan Provincial Tumor Hospital,Kunming 650118,China)
OBJECTIVE:To establish a method for the concentration determination of indigo in rats’plasma,and study the pharmacokinetic characteristics in rats in vivo.METHODS:18 rats were randomly divided into low-dose,medium-dose,high-dose*主管药师,硕士。研究方向:临床药物治疗、药事管理。电话:0871-68217128。E-mail:vampireicecream@163.com#通信作者:主治医师。研究方向:肿瘤危重症治疗。电话:0871-68185656-2036。E-mail:liyw20008@163.comgroups,6 in each group,which were intragastrically administrated 10,20,40 mg/kg of indigo solution.The sample blood 0.3 mL was taken from eye socket before administration and 0.083,0.25,0.5,0.75,1,2,4,6,8,10,12,16,24,48,72 h after administration,separating the plasma,then LC-MS/MS was used to determine the plasma concentration of indigo after methanol precipitation.The column was Agilent Poroshell EC-C18with mobile phase consisting of methanol-5 mmol/L ammonium acetate solution(95∶5,V/V)at a flow rate of 0.4 mL/min;multiple reaction monitoring was conducted for the quantitative analysis,with ion pair of 263.1-218.8(indigo)and 237.2-194.1(carbamazepine,internal standard).Pharmacokinetic parameters were calculated by DAS 3.0 software.RESULTS:The linear range of indigo was 0.5-100 ng/mL(r=0.999 9),intra-day and inter-day RSDs were lower than 9%(n=5);matrix effects of low,medium and high does quality control samples were(98.25±3.71)%,(102.23± 2.64)%,(102.29±3.79)%(n=5).The pharmacokinetic parameters of indigo in low-dose,medium-dose,high-dose groups were tmaxof(8.6±1.1),(9.2±0.8)and(9.5±0.8)h;cmaxof(30.9±8.6),(44.9±10.1),(96.1±17.4)ng/mL;t1/2of(14.9±2.1),(16.3±2.9),(15.3±3.7)h;AUC0-72hof(366.6±83.4),(694.9±105.8),(1 223.42±108.7)ng·h/mL,respectively.CONCLUSIONS:The method shows high sensitivity,good specificity,and can be used for the content determination of indigo in plasma samples of rats.The pharmacokinetics of indigo in rats in vivo fits non-compartment model.
Indigo;LC-MS/MS;Rats;Plasma;Pharmacokinetics
R965
A
1001-0408(2017)07-0912-04
2016-07-25
2016-12-19)
DOI10.6039/j.issn.1001-0408.2017.07.14
