钠离子电池正极材料研究进展
2017-03-13方永进陈重学艾新平杨汉西曹余良
方永进 陈重学 艾新平 杨汉西 曹余良,*
(1武汉大学化学与分子科学学院,武汉430072;2武汉大学动力与机械学院,武汉430072)
钠离子电池正极材料研究进展
方永进1陈重学2艾新平1杨汉西1曹余良1,*
(1武汉大学化学与分子科学学院,武汉430072;2武汉大学动力与机械学院,武汉430072)
近年来,钠离子电池由于资源丰富、价格低廉等特点,逐渐成为储能领域的研究热点。然而,钠离子具有较大的离子半径和较慢的动力学速率,成为制约储钠材料发展的主要因素,而发展高性能的嵌钠正极材料是提高钠离子电池比能量和推进其应用的关键。本文详细综述了目前钠离子电池研究的正极材料体系,包括过渡金属氧化物、聚阴离子类材料、普鲁士蓝类化合物、有机分子和聚合物、非晶材料等,并结合这几年我们课题组在正极方面的研究工作,探讨了材料的结构和电化学性能的关系,分析了提高正极材料可逆容量、电压、结构稳定性的可能途径,为钠离子电池电极材料的发展提供参考。
钠离子电池;正极材料;研究进展;储钠反应;电化学反应机理
1 引言
能源和环境问题是当今世界最受关注的议题。目前,化石能源(煤、石油和天然气等)每年的消耗量占全球能源总消耗量的85%以上,而化石能源储量有限,人类对能源的需求增长和对化石能源无节制地开采,必将导致化石能源枯竭的窘境。同时,化石能源的使用会引发环境污染、温室效应、雾霾等环境问题,因此急需探索新的能源体系。可再生能源具有天然的自我再生功能,是人类取之不尽用之不竭的能源。可再生能源包括太阳能、风能、潮汐能、水能、地热能、海洋能、生物质能等,但由于这些新能源体系具有很强的地域性和间歇性,使得其有效的利用面临着许多技术问题,而规模储能是解决这些难题的关键技术。在各种储能方式中,电化学储能是最为简便、高效的一种方式,成为储能技术发展的主流。其中锂离子电池因其具有能量密度高、工作电压高、循环寿命长、自放电率小和绿色环保等突出优势,在手机、笔记本电脑、数码相机、电动工具等领域得到了广泛应用,并逐步向新能源汽车和储能等领域拓展。

方永进,2011年本科毕业于湖北大学化学工程与工艺专业,2016年博士毕业于武汉大学物理化学专业。主要研究方向为新型钠离子电池电极材料。

艾新平,现为武汉大学化学与分子科学学院教授、博士生导师、国家863节能与新能源汽车重大专项立项评审与监理专家。主要研究领域为电化学能源材料及技术,如锂硫电池、硅负极材料等。

杨汉西,现为武汉大学化学与分子科学学院教授,博士生导师,从事电化学能源转换领域新材料、新技术和新体系的应用基础研究,在锂/钠离子电池等化学电源方面有重要影响。

曹余良,2003年获得武汉大学博士学位,2009-2011年,美国西北太平洋国家实验室访问学者。现为武汉大学化学与分子科学学院教授、博士生导师、教育部新世纪优秀人才。主要研究领域为钠离子电池电极材料和电解液相关研究。
然而,不断增长的锂离子电池市场,必然带来锂资源短缺和锂价格上涨的问题。若将每kWh锂离子电池(三元氧化物正极)用锂量折合以碳酸锂计约为0.65 kg,按目前年产5000万辆汽车均配备10 kWh电池计算,则每年至少需要32万吨碳酸锂,仅此一项碳酸锂的需求量将是目前开采量的~1.7倍;如考虑到纯电动汽车,每车至少配备大于40 kWh的电池,用锂量将更加巨大1。此外,从大规模储能应用角度考虑,2014年全球新能源装机达6.53亿千瓦(包括4.64亿千瓦风电和1.89亿千瓦光伏),如果配备储能系统去提高新能源利用效率,按一天10小时计算,累计需储能容量为65.3亿千瓦时(6530 GWh)2。而就目前全球锂的资源储量(以碳酸锂当量计算约为7100万吨),按照每千瓦时能量消耗0.65 kg碳酸锂计算,仅能够满足1092亿千瓦时(109200 GWh)的能量。计算结果显示,锂资源的稀缺使得锂离子电池难以同时支撑电动汽车和大规模储能两大产业的发展。由此,需要发展资源丰富和价格低廉的新型储能体系。
钠离子电池体系由于具有资源丰富、价格低廉、环境友好,以及与锂离子电池相近的电化学性质,近几年受到广泛关注,为电化学储能提供了新的选择。钠元素在地壳中含量排第六位(2.75%),具有非常高的丰度,且分布广泛。同时,钠与锂具有相似的物理化学性质,钠离子电池与锂离子电池也具有相似的电化学反应机制,因此钠离子电池有望表现出与锂离子电池相当的电化学性能。钠离子电池结构和原理与锂离子电池基本相同(如图1所示),正负极选用具有不同电势的钠离子嵌入化合物,电解液选用钠盐的有机电解液。充电时,Na+从正极脱出经过电解液嵌入负极,同时电子经过外电路流入负极以保持电荷平衡。放电过程正好相反,Na+由负极脱出嵌入正极,电子经外电路流入正极。从嵌钠电化学合金反应看,钠离子电池在成本上的另一个优势是钠元素不会与铝集流体发生合金化反应,使得负极也可以用便宜的铝替代铜(锂离子电池负极集流体)作为集流体,进一步降低了体系成本。因而,钠离子电池在储能领域表现出非常广阔的应用前景。
然而由于钠离子比锂离子具有更大的离子半径,使得钠离子电池相比锂离子电池具有更迟缓的扩散动力学,这给探寻合适嵌钠正负极材料提出了挑战。经过近些年的研究,一些颇具应用前景的高性能材料体系不断涌现,如过渡金属氧化物、磷酸盐、普鲁士蓝类化合物、碳基材料、合金类负极、转换反应材料等。从目前的研究看,正极材料的比容量(80-150 mAh·g-1)仍远远低于负极材料(碳材料:~250 mAh·g-1;合金材料:400-600 mAh·g-1),因此发展高性能的嵌钠正极材料是提高钠离子电池比能量和推进其应用的关键。本文着重围绕目前所研究的嵌钠正极材料体系,并结合近几年我们课题组在正极材料方面的研究工作,综述了钠离子电池正极材料的研究进展,详细介绍了其结构、性能和制备技术,探讨了提高嵌钠正极材料的容量、电压和结构稳定性的可能途径,阐述其发展趋势及应用前景。

图1 钠离子电池反应原理图Fig.1 Working principle of sodium ion batteries
2 钠离子电池正极材料
钠离子电池的电化学性能主要取决于电极材料的结构和性能,通常认为,正极材料的性能(如比容量、电压和循环性)是影响钠离子电池的能量密度、安全性以及循环寿命的关键因素。因此,正极材料性能的改善和提升,以及新型正极材料的开发和探索一直是钠离子电池领域的研究热点。储钠正极材料的选取原则为:(1)具有较高的比容量;(2)较高的氧化还原电位;(3)合适的隧道结构,有利于钠离子嵌入脱出;(4)良好的结构稳定性;(5)良好的电子和离子导电率;(6)价格廉价、资源丰富、环境友好等。
2.1 钠离子电池正极材料的发展
实际上,储钠电极的研究可以追溯到20世纪70年代,几乎与储锂材料的研究齐头并进,主要原因是它们具有相近的原子结构和电化学特征。早期储钠正极材料的研究主要集中在过渡金属氧化物材料体系。1980年,Goodenough等3报道了LiCoO2层状氧化物嵌脱锂的电化学性能。同一年,Hagenmuller等4报道了四种不同结构的层状NaxCoO2的储钠性能,四种结构分别为:0.55≤x≤0.60(P′3);0.64≤x≤0.74(P2);x=0.77(O′3)和x=1(O3)。1982年,Abraham5发表了插层材料储钠性能的综述文章。1982年,Hagenmuller等6报道了NaCrO2与NaNiO2可以实现0.15 Na的嵌脱反应。1985年,Hagenmuller等7研究了NaxMnO2的储钠性能。但是自从1990年后,锂离子电池的实用化吸引了人们的注意力,而本身在能量密度指标上并不占优势的钠离子电池的研究则在随后的20年中几乎处于停滞状态8。近年来,得益于新能源对储能电池的应用需求,人们又一次将目光转向更具资源优势的钠离子电池,开发出一系列储钠电池材料。对于正极材料来说,主要包括过渡金属氧化物材料、聚阴离子类材料、普鲁士蓝类材料、有机分子和聚合物、非晶材料等,它们的实际比容量与电压图的关系如图2所示。下面就正极材料的研究进展进行具体介绍。
2.2 过渡金属氧化物材料
过渡金属氧化物可以用NaxMeO2表示,其中Me为过渡金属,包括Mn、Fe、Ni、Co、V、Cu、Cr等元素中的一种或几种;x为钠的化学计量数,范围为0

图2 钠离子电池正极材料实际容量与电压图Fig.2 Relationship between capacity and voltage for cathode materials in sodium ion batteries
氧化物的电化学性能由相结构的特点所决定,而相结构又与原始态的钠含量、层的稳定性、钠原子周围的环境等因素相关。同时由于钠离子半径较大,在电化学过程中钠离子的迁移势必会造成氧层的滑移,常常伴随着一系列相变的发生,如O3―P3或O2―P2的相转变(图3)。这些相转变一方面存在能垒,影响离子在体相的扩散;另一方面复合的相变过程存在较大的结构变化,造成循环过程结构的瓦解,影响循环性能。通常对于O3结构的氧化物(NaMeO2)来说,由于其具有更多的嵌钠位点,因此具有较高的容量。而P2相结构(Na0.67MeO2)具有更大的层间距,使得Na+扩散较为容易,可以从一个三棱柱空位迁移到邻近的一个三棱柱空位,表现出更高的离子电导率。因此,在储钠层状氧化物材料的研究中,主要工作集中于材料体相元素掺杂或取代,以此来减弱相转变,提高材料的结构稳定性。
而当氧化物中钠含量较低时(x<0.5),主要以三维隧道结构的氧化物为主(Na0.44MnO2等),隧道型氧化物具有独特的S型和五角形隧道(图3),具有稳定的结构,在空气中可以稳定存在,充放电过程中性能稳定,但是这种材料首周充电容量较低,后面将作详细介绍。

图3 隧道、O3、P3、O2、P2相过渡金属氧化物的晶体结构示意图Fig.3 Crystal structure illustration of tunnel-type oxide and layered oxides with O3,P3,O2,P2 structures
下面,从不同的活性元素角度介绍一下过渡金属氧化物的研究进展。
2.2.1 锰基氧化物
2.2.1.1 隧道型氧化物
Na0.44MnO2是一种典型的隧道型储钠材料,它属正交晶系,空间群为Pbam,主要由MnO5四棱锥和MnO6八面体构成S型和五角形的隧道,三种不同位点的钠处于隧道中(图3)。Doeff等11首先研究了Na0.44MnO2在固体聚合物电解质中的储钠性能,材料具有可逆的储钠性能,但循环性能较差。Sauvage等12通过非现场 XRD研究了Na0.44MnO2的储钠机制,发现在2.0-3.8 V充放电过程中存在六个两相反应区间,但可逆容量和循环性能都较差。Cao等13通过聚合物热解法合成了Na0.44MnO2纳米线,首次实现了Na0.44MnO2的高稳定长寿命循环(循环1000周容量保持率为77%)(图4)。该材料的可逆比容量高达128 mAh·g-1,这一超出理论比容量的现象可能归于五角形隧道的储钠。Kim等14通过DFT的方法研究了Na0.44MnO2的结构和电化学特性,发现在脱钠产物Na0.22MnO2的结构中,S型的隧道仍然存在部分钠离子,同时一些中间相和两相反应也被验证。随后,不同方法合成的各种形貌的Na0.44MnO2材料都得到广泛的报道15。Guo等16通过Ti的取代得到Na0.61Ti0.48Mn0.52O2隧道结构材料,该材料表现出2.9 V的平均电压和86 mAh·g-1的可逆比容量。Jiang等17合成了Na0.54Mn0.50Ti0.51O2/C材料,具有137 mAh·g-1的可逆容量,循环400周容量保持率在85%以上。Hu等18探索了NaxMn1-yTiyO2(x=0.44,0.66)在水溶液电解液中的电化学性能,相对于层状氧化物材料对水分敏感,隧道型的NaxMn1-yTiyO2在水溶液电解液中表现出稳定的电化学性能。例如,Na0.44Mn0.44Ti0.56O218(a)在水溶液中具有38 mAh·g-1的可逆容量,并且稳定循环400周。Hu等19合成了对空气稳定的Na0.61Mn0.27Fe0.34Ti0.39O2材料,具有90 mAh·g-1的可逆比容量和3.56 V的工作电压,对应着Fe3+/4+的氧化还原,同时,他们采用现场X射线衍射谱(XRD)、现场X射线吸收谱和非现场穆斯堡尔谱分析了该材料的储钠反应机制。
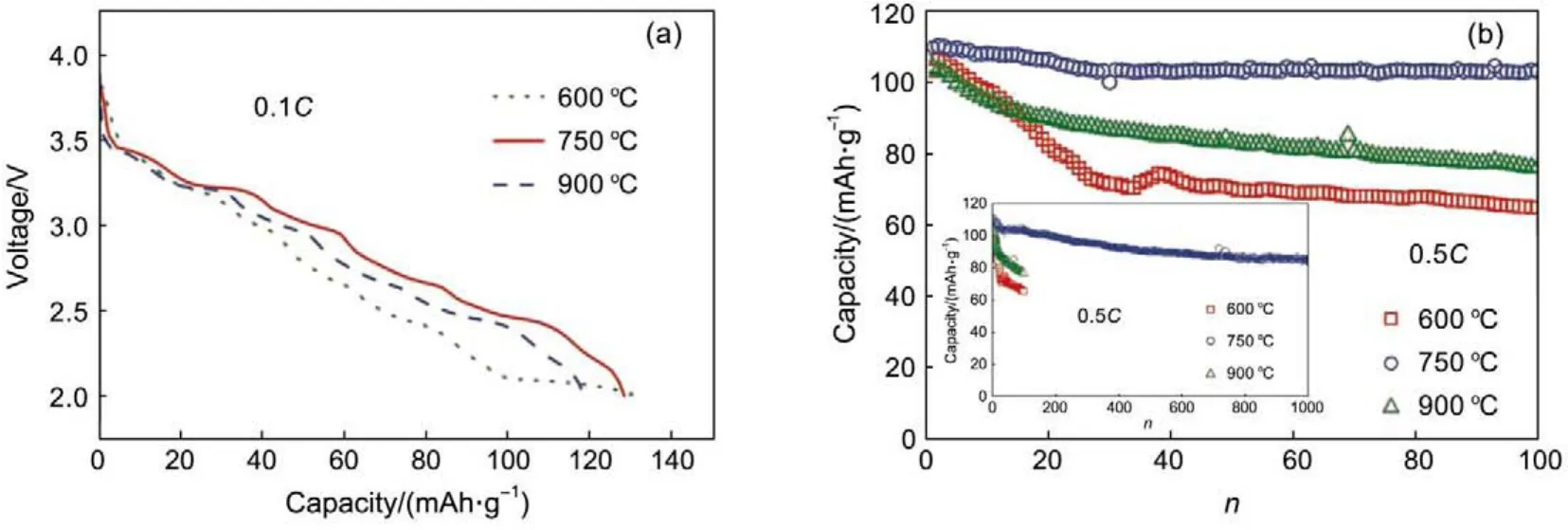
图4 Na0.44MnO2纳米线的电化学性能13Fig.4 Electrochemical performance of Na0.44MnO2nano wire13
总体来说,相对层状氧化物,这种隧道结构的氧化物由于存在Mn-O八面体的相互支撑,钠离子在嵌入脱出过程中,材料结构仍然能够保持相对稳定,这大大提高了材料的循环稳定性。然而,这种材料初始钠含量过低,造成可逆容量较低。因此,提高材料钠含量,并保持稳定的隧道结构是这类材料的发展方向。

图5 不同结构NaxMnO2的充放电曲线7Fig.5 Charge and discharge curves of different NaxMnO2structures7
2.2.1.2 层状氧化物
Mn基的层状氧化物因合成条件和化学计量比不同,主要表现为三类:P2-Na0.7MnO2+y,单斜的O′3型α-NaMnO2和正交P2型的β-NaMnO2(图5)。早在1985年,Mendiboure等7就系统地研究了这三种化合物的储钠性质。2011年,Ma等20重新研究了O′3-NaMnO2的电化学性能,在2.0-3.8 V的充放电区间可以获得185 mAh·g-1的可逆容量,但是容量衰减较快。充放电曲线表现出一系列的阶梯,对应着多步的结构转变。Abakumov等21通过透射电镜、同步辐射XRD并结合理论计算等,发现α-NaMnO2存在大量平面缺陷,这些缺陷会影响材料的结构、磁性和电化学性能,通过计算预测高浓度的缺陷会引起α-NaMnO2向β-NaMnO2的畸变。
相对α-和β-NaMnO2,P2-NaxMnO2(x=0.7)表现出更优异的电化学性能。Caballero等22较早研究了P2-NaxMnO2的相关性质,发现水分子可以进入钠的层间,使层间距增加0.25 nm。无水的P2-NaxMnO2材料可以给出140 mAh·g-1的可逆比容量,经过几周的充放电后,晶体结构会慢慢坍塌变成无定型结构。NaxMnO2结构的扭曲是由高自旋的Mn3+离子的姜-泰勒效应引起的,通过Al3+、Li+、Mg2+、Ni2+、Co3+等掺杂或取代可以抑制姜-泰勒效应,从而获得结构稳定的P2相材料23。Yuan等24在P2型氧化物中引入惰性Al3+离子掺杂合成了Na0.67[Mn0.65Ni0.15Co0.15Al0.05]O2,结果显示Al掺杂材料的首周可逆容量虽比未掺杂的稍低,但是Al掺杂材料表现出更稳定的电压曲线行为,循环50周,容量几乎没有衰减。同时,掺杂材料也表现出非常好的倍率性能(图6)。Yabuuchi等25也通过Li+取代得到P2-Na5/6[Li1/4Mn3/4]O2材料,Li与Mn在过渡金属层中有序排列,而Na则均匀、无序地分布在共边和共面的三棱柱位置。该材料具有200 mAh·g-1的可逆比容量,同时充放电机制转为固溶体反应,充放电曲线变得平滑。Mg2+掺杂也可以有效的抑制姜-泰勒效应引起的变形,主要原因可能是提高了Mn4+浓度。Billaud等26研究了不同Mg掺杂量对Na0.67Mn1-xMgxO2(0≤x≤0.2)材料电化学性能的影响,发现Mg掺杂可以提高材料的稳定性,5%的Mg掺杂材料具有175 mAh·g-1的可逆比容量,提高Mg的掺杂量,材料可逆容量降低。最近,Komaba小组27报道的Mg掺杂的Na0.67Mg0.28Mn0.72O2材料,可以获得超过理论200 mAh·g-1的比容量,增加的容量可能来源于氧的可逆氧化和还原。
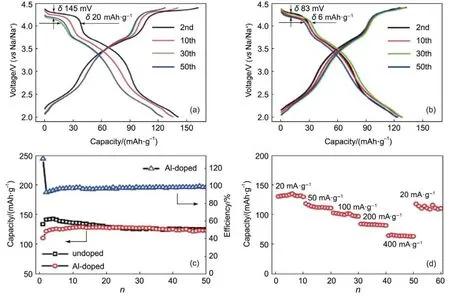
图6 Na0.67[Mn0.65Ni0.15Co0.2]O2和Na0.67[Mn0.65Ni0.15Co0.15Al0.05]O2材料的电化学性能24Fig.6 Electrochemical performances of Na0.67[Mn0.65Ni0.15Co0.2]O2and Na0.67[Mn0.65Ni0.15Co0.15Al0.05]O2materials24
Billaud等28重新研究了β-NaMnO2的电化学性能,可以获得190 mAh·g-1的可逆比容量,在2C的倍率下可以获得142 mAh·g-1的容量,同时循环100周容量保持率为70%。通过X射线衍射(XRD),高分辨透射电镜(HRTEM)和核磁共振谱(NMR)对反应机制进行了分析,发现反应过程中存在复杂的变化,出现了α-NaMnO2和β-NaMnO2共存的区域。
从以上结果来看,单一Mn元素形成的层状氧化物材料实际上其电化学性能都会受到结构变化或相转变的影响,表现出较差的结构稳定性。通过活性或惰性元素掺杂或取代能够明显抑制钠脱嵌过程中的相转变,提高材料的循环稳定性。
2.2.2 铁基过渡金属氧化物
α-NaFeO2属三方晶系,空间群为R3m,是典型的O3结构层状材料。NaFeO2的电化学测试表明,该材料表现出3.3 V左右的放电平台,并且电池性能与截止电压相关,当充电截止电压超过3.5 V时,由于钠脱出量过多导致Fe4+迁移至钠层而引起结构的不可逆变化,导致可逆放电容量降低。当截止电压在3.5 V时,该材料可以给出100 mAh· g-1的可逆容量,对应着~0.3 Na的可逆脱嵌29(图7)。Zhao等30通过示差扫描量热法(DSC)研究发现,全充态的Na0.58FeO2在高于300°C时会发生分解。
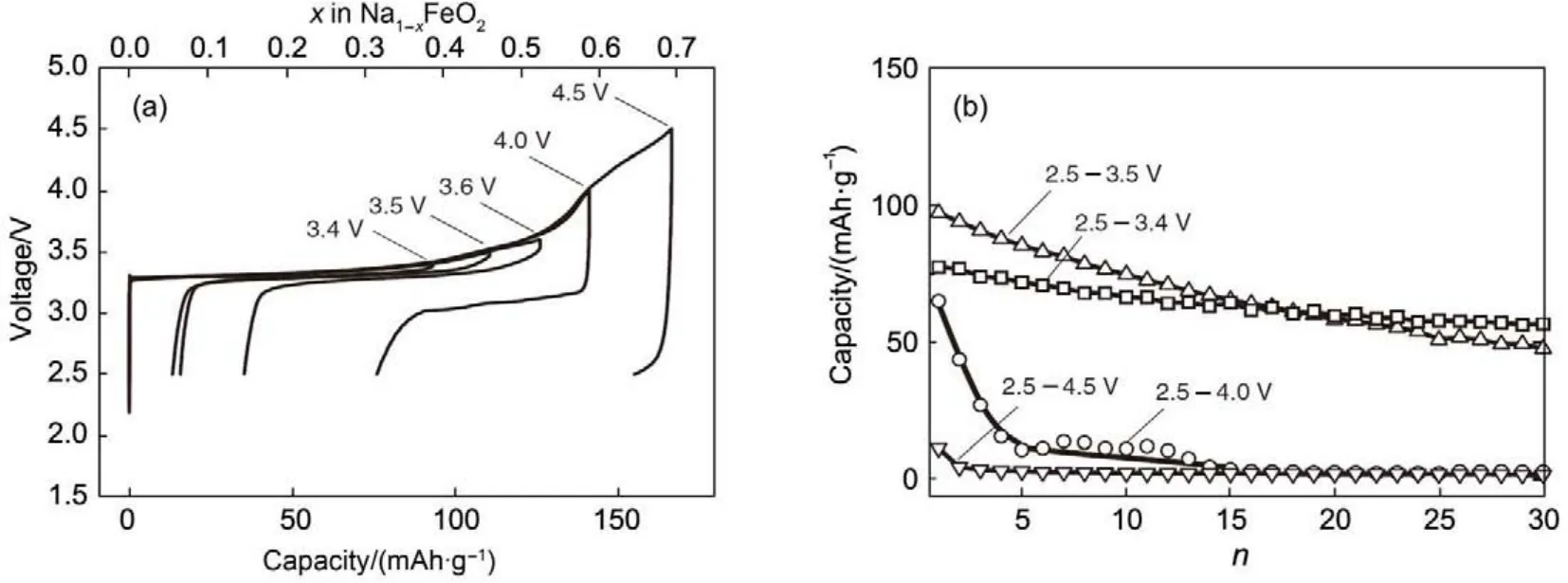
图7 NaFeO2在不同截止电压下的电化学性能29Fig.7 Electrochemical performance of NaFeO2tested at varied cut-off potential29
Lee等31通过非现场的穆斯堡尔谱和现场的同步辐射XRD研究了导致α-NaFeO2电化学不可逆的原因,发现充电后的Na1-xFeO2电极在开路状态下,超过20%的Fe4+会自发的还原为Fe3+,这种自放电行为会导致电解液的分解,从而引起电极阻抗的增加和效率的降低。现场同步辐射研究了NaFeO2电极的非平衡相转变行为,发现脱钠过程中有一个新的Oʺ3相的存在,循环过程中,出现了不对称的结构变化。
为了提高NaFeO2电极的稳定性,并获得高的工作电压和比能量,可以采用Mn、Ni、Co等元素对Fe进行取代,从而改善其电化学性能。从资源和成本考虑,选择Mn取代应更具有优势,同时可以获得比较高的容量和稳定性。Komaba等32比较了O3-NaFe1/2Mn1/2O2和P2-Na2/3Fe1/2Mn1/2O2电化学性能的区别,发现O3相在4.3-1.5 V可以放出110 mAh·g-1的容量,而P2相则可实现高达190 mAh· g-1的可逆比容量,其中Fe和Mn都为活性元素,发生Mn3+/Mn4+和Fe3+/Fe4+的氧化还原,两种材料的循环稳定性相近。然而这种Fe-Mn基层状氧化物材料存在的主要问题是:(1)α-NaFeO2充电高于3.4 V后的不可逆相转变,影响材料循环性;(2)Mn2+/3+/4+的低氧化还原电势,拉低了材料的平均电压;(3)二价锰离子在电解液中的溶解流失。通过降低材料中活性较差的铁元素的含量和略微添加高氧化还原电位的镍元素,可以明显改善材料的循环稳定性和平均电压。Yuan等33制备了P2-Na0.67Mn0.65Fe0.35O2和Na0.67Mn0.65Fe0.2Ni0.15O2两种化合物(如图8所示),两种化合物都可以给出超过200 mAh·g-1的初次比容量,其中Ni取代的化合物循环50周,容量保持率为71%,表现出较好的稳定性,主要原因为Ni的取代可以提高材料的稳定性并缓和Mn(III)引起的姜-泰勒效应。Thorne等34研究了一系列的NaxFexMn1-xO2(0.5
镍取代的铁基氧化物也得到研究,O3-NaFe1-xNixO2(0.5≤x≤0.7)在2.0-3.8 V之间,当x=0.5时,可以获得112 mAh·g-1的可逆比容量和89%的首周效率,平均电压为2.85 V,x=0.7时,可以获得135 mAh·g-1的比容量,首周效率为93%,平均电压为2.7 V(图9)。Ni取代后,Ni3+/Ni4+的氧化还原可以抑制Fe4+的姜-泰勒效应,从而可以减小充放电极化36。最近,Ti取代的NaFex(Ni0.5Ti0.5)1-xO2(x=0.2和0.4)可以表现出高于3.1 V的放电电压和120 mAh·g-1的可逆容量,充放电曲线也变得平滑37。

图8 P2-Na0.67Mn0.65Fe0.35O2和P2-Na0.67Mn0.65Fe0.2Ni0.15O2样品的电化学性能33Fig.8 Electrochemical performances of P2-Na0.67Mn0.65Fe0.35O2and P2-Na0.67Mn0.65Fe0.2Ni0.15O2samples33
2.2.3 镍基过渡金属氧化物
单斜晶系的NaNiO2,属于C2/m空间群,为O′3相结构。由于Ni3+具有姜-泰勒效应,使得晶胞的a/b值由1.73变到1.866,38。1982年Hagenmuller等6研究发现NaNiO2在3.5-2.0 V可以实现0.2 Na的脱出,充放电曲线包含多个平台,对应着不同的相变。Ceder等39重新研究了O′3-NaNiO2的电化学性能,由于较小的Ni3+不会迁移到Na的位置,因此材料表现出较好的电化学性能。如图10a所示,这种化合物在1.25-3.75 V电压范围,首周的充放电比容量分别为147/123 mAh·g-1(0.63/0.52 Na),充放电曲线都由一系列的平台组成,循环20周后依然有116 mAh·g-1的比容量,表现出较好的循环稳定性。在4.5-2.0 V电压范围,可以获得146 mAh·g-1的可逆容量,但由于在高电压下结构遭受严重破坏,循环性能变差。Han等40通过现场XRD研究了NaNiO2首周的电化学性能和结构演变,其结构变化顺序为Oʹ3-Pʹ3-Pʺ3-Oʺ3-Oʹʺ3,分别对应着NaNiO2、Na0.91NiO2、Na0.84NiO2、Na0.81NiO2和Na0.79NiO2的相互转变,这些晶相的主要区别为堆积层错的不同(图10)。

图9 NaFeO2、NaFe0.5Ni0.5O2、NaFe0.3Ni0.7O2样品的充放电曲线36Fig.9 Charge/discharge curves of NaFeO2,NaFe0.5Ni0.5O2, and NaFe0.3Ni0.7O2samples36
Ni3+的姜-泰勒效应,以及充放电过程的相转变、Na重新排布,都会引起NaNiO2结构的不稳定,通过元素取代可以进一步提高其电化学性能。虽然NaNiO2中Ni价态为+3,但是经Mn或Ti元素取代后价态一般呈现为+2,Ni元素可实现+2↔+3↔+4价的氧化还原,而Mn或Ti元素保持+4价,提高了材料的稳定性。如图11,O3-NaNi0.5Mn0.5O2材料的充放电曲线表现出很多的台阶,而P2-NaxNi1/3Mn2/3O2材料拥有平滑的充放电曲线41。非现场XRD研究表明,O3-NaNi0.5Mn0.5O2材料充电过程中存在着O3-Oʹ3-P3-Pʹ3-Pʺ3的变化,表明循环过程中存在着许多平面的滑移和强的姜-泰勒效应。由于O3相材料含有较多的钠,使得它具有更高的可逆容量,但是P2相表现出更好的稳定性。
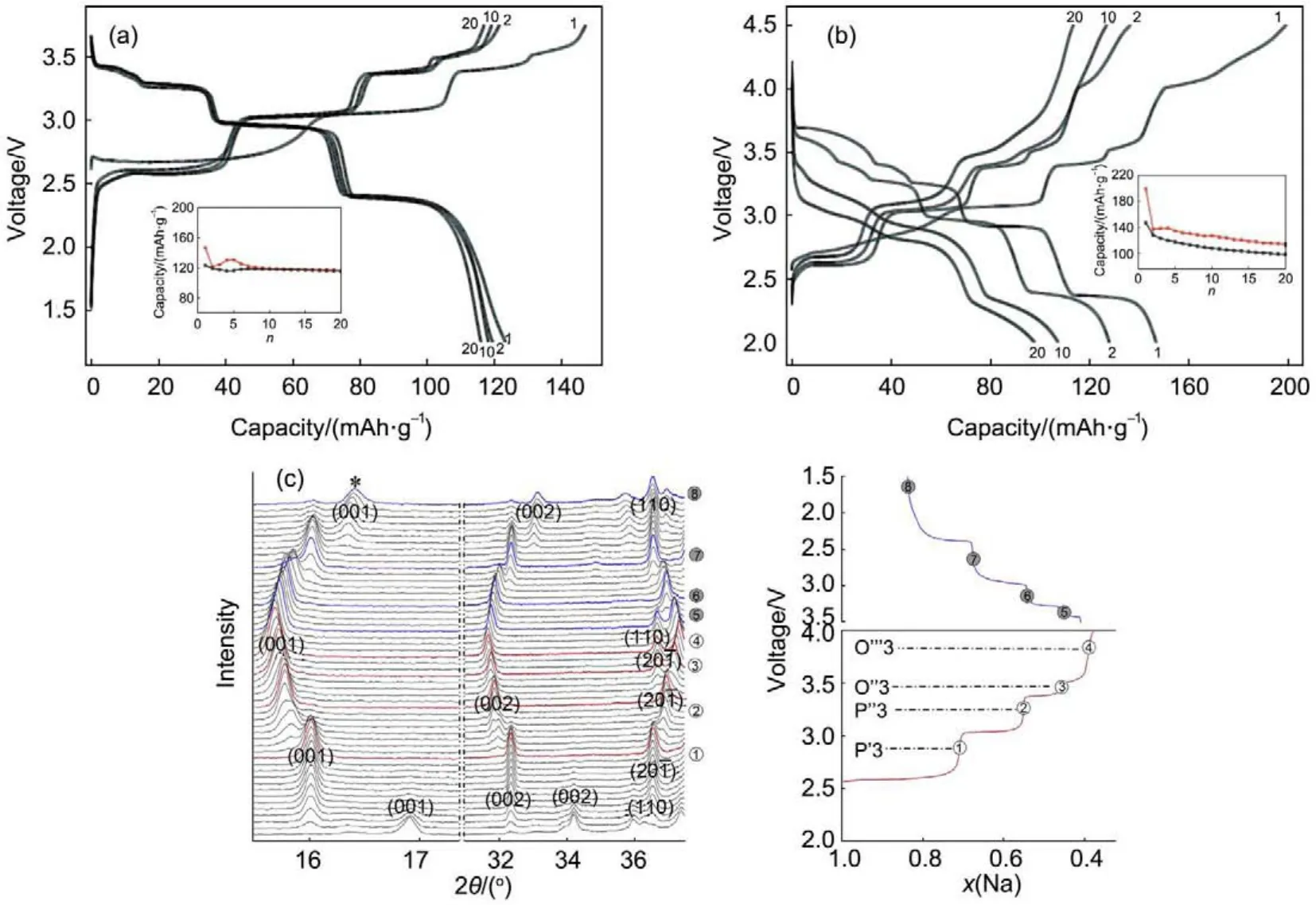
图10 Oʹ3-NaNiO2的充放电曲线和结构演化40Fig.10 Charge-discharge curves and structural evolution of Oʹ3-NaNiO240

图11 O3-NaNi0.5Mn0.5O2和P2-Na2/3Ni1/3Mn2/3O2充放电曲线41Fig.11 Charge/discharge curves of(a)O3-NaNi0.5Mn0.5O2and(b)P2-Na2/3Ni1/3Mn2/3O241
Yuan等42通过溶胶凝胶法合成了一系列Fe取代的O3-NaFex(Ni0.5Mn0.5)1-xO2(x=0,0.1,0.2,0.3, 0.4),发现Fe取代可以抑制Na空位的有序性,对NaFex(Ni0.5Mn0.5)1-xO2的可逆容量、循环性能和倍率性能均有很大的提高。其中,NaFe0.2(Ni0.5Mn0.5)0.8O2(x=0.2)的材料具有131 mAh·g-1的可逆容量,循环30周容量保持率为95%,表现出较好的稳定性,同时该材料具有10C的倍率性能(图12)。这些性能远远高于未取代的O3-NaNi0.5Mn0.5O2材料,表明Fe取代可以平滑相变,提高可逆容量和循环稳定性。同时,他们对反应过程中的结构演化进行了详细分析,发现NaNi0.5Mn0.5O2材料充电过程经历O3-P3-P3ʺ的相变过程,而NaFe0.2(Ni0.5Mn0.5)0.8O2材料却经历O3-P3-OP2的相变,不同的反应机理使得Fe取代的材料在高电压区间稳定性增强。
Ti取代的P2-Na2/3Ni1/3Mn1/2Ti1/6O2材料在2.5-4.5 V之间可以给出127 mAh·g-1的可逆比容量,Ti的取代可以提高材料结构稳定性,但是由于Ti没有电化学活性,掺入后将降低可逆容量43。O3-NaNi0.5Ti0.5O2材料在2.0-4.7 V之间,首周的充放电比容量为170/121 mAh·g-1,但50周后容量衰减至52.8%,当限制电压区间为2.0-4.0 V时,材料循环100周容量保持率为93.2%,表现出极好的循环稳定性44。P2-Na2/3Ni1/3Ti2/3O2属于六方晶系,P63/ mmc空间群。Shanmugam等45研究发现,这一化合物在不同的电压范围可以分别实现Ni2+/Ni3+和Ti4+/ Ti3+的氧化还原,既可以作为正极也可以作负极。在4.2-2.0 V电压区间,放电中压为3.7 V,容量为75 mAh·g-1,可作为正极;在2.0-0.2 V电压区间,具有75 mAh·g-1的比容量,放电电压为0.7 V,可作为负极使用。最近,Te和Sr取代的化合物 Na2Ni2TeO6和 Na1.6Sr0.2Ni2TeO6也 被 研 究46,Na2Ni2TeO6在3.0-4.35 V间可以给出110 mAh·g-1的可逆比容量,而Na1.6Sr0.2Ni2TeO6也具有108.5 mAh·g-1的可逆比容量,两类材料都具有约3.7 V的放电平台,且稳定性较好。钴的取代也在较早被研究,Saadoune等47以O3-NaNi0.6Co0.4O2为原料通过化学脱钠法制备了P3-Na0.58Ni0.6Co0.4O2材料,并研究了嵌钠过程中结构的变化,发现随着钠离子嵌入,该材料发生P3-Pʹ3-Oʹ3-O3的相变,整个反应可以嵌入0.4Na。Johnson课题组48报道了Li取代的P2-Na0.85Li0.17Ni0.21Mn0.64O2材料,得到3.4 V的放电电压和100 mAh·g-1的可逆容量及较好的倍率性能。Li的取代可以稳定过渡金属层,平滑充放电曲线。同时,随着Li含量的增加,材料逐渐从Na-O3(O2-NaxLixNizMn1-y-zO2,x>y)转变为Na-P2(P2-NaxLixNizMn1-y-zO2,x > y), 再 到 Li-O3(O2-LixNaxNizMn1-y-zO2,x>y),选择适当的Li含量,可以得到两相或者三相共存的材料(图13)。Lee49(b)和Guo49(a)等分别报道了 P2+O3共存的化合物Na0.66Li0.18Mn0.71Ni0.21Co0.08O2+σ和Na0.7Li0.3Ni0.5Mn0.5O2,这类材料结合了P2相倍率性能较好和O3相比容量较高的优点,循环过程更加稳定。

图12 (a)O3-NaNi0.5Mn0.5O2和(b)O3-NaFe0.2(Ni0.5Mn0.5)0.8O2充放电曲线42Fig.12 Charge/discharge curves of(a)O3-NaNi0.5Mn0.5O2and(b)O3-NaFe0.2(Ni0.5Mn0.5)0.8O242

图13 Na1-xLixNi0.5Mn0.5O2+σ随x的变化的XRD图49(b)Fig.13 X-ray diffraction(XRD)patterns of Na1-xLixNi0.5Mn0.5O2+σwith different Li contents(x)49(b)
通过元素掺杂取代一方面可以获得嵌钠稳定的晶体结构,另一方面可以改善嵌脱机理而展现不同的储钠性能。掺杂元素多见于+1,+2,+3和+4价元素。实际上,在层状氧化物中掺入+5和+6价元素并非不可能,只是这类化合物过去研究得较少。如果层状AMO2氧化物中恰好1/3 M被X所替代时,分子式就可以写成A3M2XO6或A2M2XO6形式(M为+2价离子;X为+5或+6价;A的浓度取决于X的价态)。此时M:X价态比为1:2,可能形成“蜂窝”有序结构,即每个XO6被六个MO6所包围。Yuan等50首次将蜂窝有序材料O3-Na3Ni2SbO6用作储钠正极,Na3Ni2SbO6电极的首周充电和放电比容量分别为122/117 mAh·g-1,相当于完全脱嵌2个Na离子,首周效率也高达95%(图14)。同时,该材料循环50周,容量保持率为95%,500周后容量保持率为70%,表现出高度的循环稳定性。该材料在30C的电流密度下,仍有90 mAh·g-1的可逆容量,表现出优异的倍率性能。同时,他们用X射线光电子能谱(XPS)、非现场XRD和非现场固体核磁谱,对材料储钠机制进行了研究,该工作为探索储钠正极材料提供了一个新方向。Ma等51随后合成有序和无序的NaNi2/3Sb1/3O2材料,详细的分析了他们的空间群、结构、晶格参数、原子排布、充放电相变等。

图14 Na3Ni2SbO6的电化学性能50Fig.14 Electrochemical performane of Na3Ni2SbO650
2.2.4 铜基过渡金属氧化物
探索新的氧化还原中心具有十分重要的意义,Hu等52报道了一系列铜基过渡金属氧化物,这些化合物中,Cu2+/Cu3+表现出电化学活性,材料表现出较好的电化学性能。例如,P2-Na0.68Cu0.34Mn0.66O2表现出67 mAh·g-1的可逆容量和3.7 V左右的放电电压52(b)。对空气稳定的P2-Na7/9Cu2/9Fe1/9Mn2/3O2材料具有90 mAh·g-1的可逆容量和3.6 V左右的放电电压,并表现出较好的循环稳定性52(c)。O3-Na0.9Cu0.22Fe0.3Mn0.48O2材料与硬碳组成的全电池具有3.2 V的平均电压和100 mAh·g-1的可逆容量52(a),并且,他们以 O3-Na0.9Cu0.22Fe0.3Mn0.48O2材料为正极,以热解无烟煤得到的碳为负极52(d),构建了一个2 Ah的电池,具有100 Wh·kg-1的实际能量密度,该电池正负极材料都使用廉价的原料,极大地降低了材料的成本。从成本和材料环境适应性(对湿气稳定)角度考虑,铜基类材料组成廉价且无毒,具有非常好的应用前景,对于设计高稳定性、低成本钠离子电池正极材料提供了一个新的方向。
2.2.5 其它过渡金属氧化物(Co,Cr,V等氧化物)
对NaxMO2(M=Co,Cr,V等)的嵌钠反应也早有研究,如在1981年,Delmas等4(b)就研究了NaxCoO2的电化学性能,他们发现随着钠的化学计量数的不同,NaxCoO2表现出不同的结构,P2(0.64≤x≤0.77)、Pʹ3(0.55≤x≤0.6)、O3(x=1)和Oʹ3 (x=0.77)结构的NaxCoO2都可以实现钠离子的嵌入和脱出,充放电曲线表现为多个平台。Berthelot等53通过现场XRD结合GITT电化学的方法,精确地研究了P2-NaxCoO2的相图,发现在0.5≤x≤1时,存在九种单相区,这些单相区的形成主要与Na-空位的有序性相关(不同钠离子排布,得到不同的单相)(图15)。Ding等54研究了P2-Na0.74CoO2的电化学性能,该材料在2.0-3.8 V之间可以给出107 mAh·g-1的可逆容量,充放电曲线的电压极化在150-250 mV,远远高于LixCoO2。非现场XRD表明,在循环过程中,c轴会膨胀和收缩,同时XPS和现场XAS证明了Co3+/Co4+的氧化还原的发生。含Na0.7CoO2的全固态电池Na15Pb4/P(EO)8NaCF3SO3/ Na0.7CoO2也较早被报道55,该电池在1.5-4.0 V区间0.5 mA·cm-2的电流密度下有1600 Wh·L-1(相对于Na)和1470 Wh·L-1(相对于Na15Pb4)的体积能量密度。
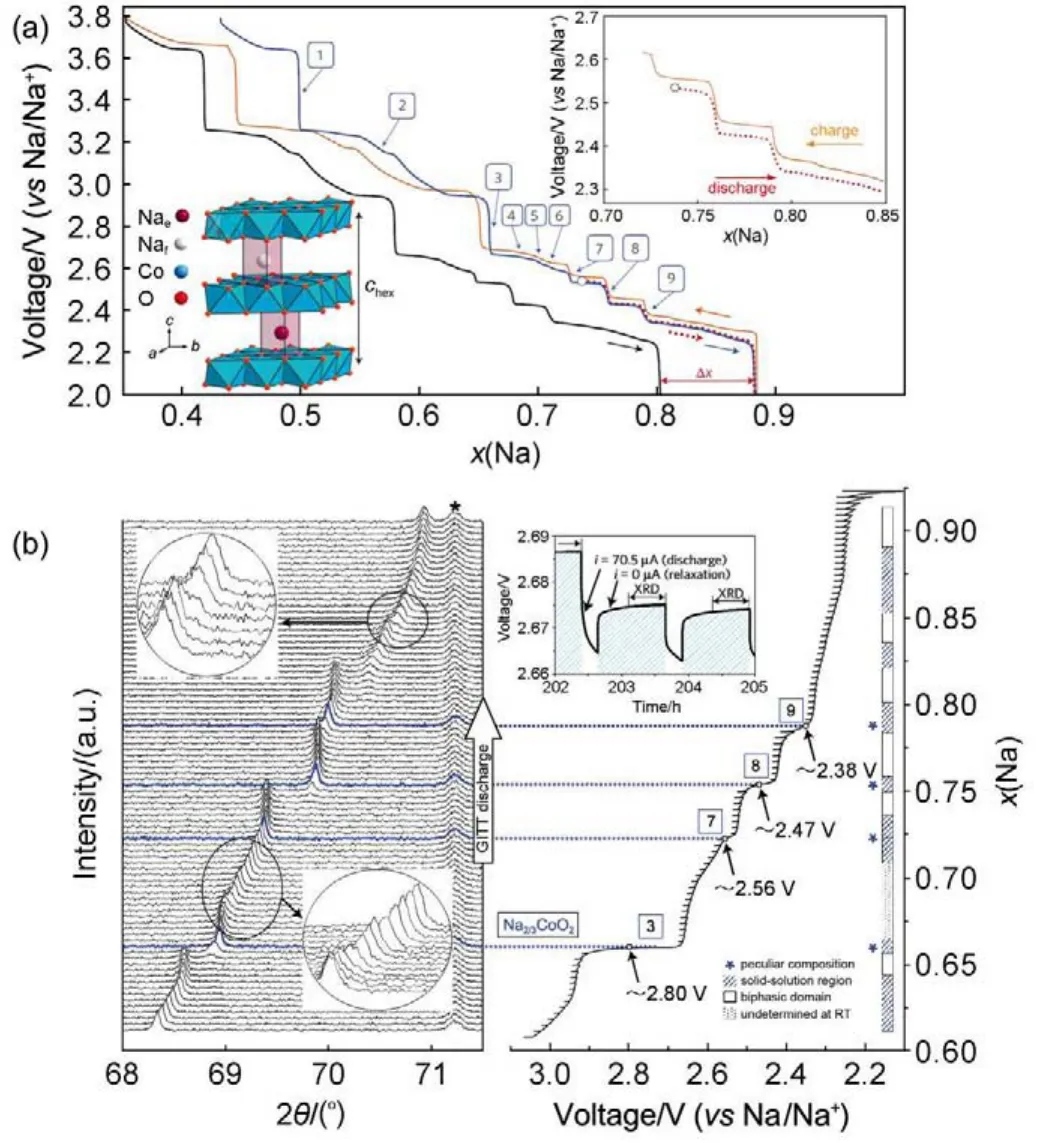
图15 P2-NaxCoO2的充放电曲线和现场XRD53Fig.15 Charge/discharge curves and structure evolution of P2-NaxCoO253
一些元素取代工作也被研究,如通过Fe取代得到O3-NaFe0.5Co0.5O2材料在2.5-4.0 V之间可以给出160 mAh·g-1的可逆容量,并可获得相对高的电压(3.14 V),同时充放电曲线也得到平滑,材料循环50周容量保持率为85%。非现场XRD也发现在充电起始阶段存在一个两相共存区(O3-P3),材料的较好倍率性能可能得益于P3相具有较快的钠离子扩散56。Carlier等57比较了Na2/3Co2/3Mn1/3O2与Na2/3CoO2电化学性能的不同。他们发现Mn取代使得充放电曲线变得平滑,说明Mn取代Co可以抑制Na-空位有序性或者MO2层的滑移。最近,P3/ P2共存的化合物也被研究58,Chen等58(b)合成了P3/ P2共存的Na0.66Co0.5Mn0.5O2化合物,在1.5-4.2 V之间,具有156.1 mAh·g-1的可逆容量,10C的倍率下循环100周容量保持率为98%,该材料具有较好的倍率性能和循环稳定性。
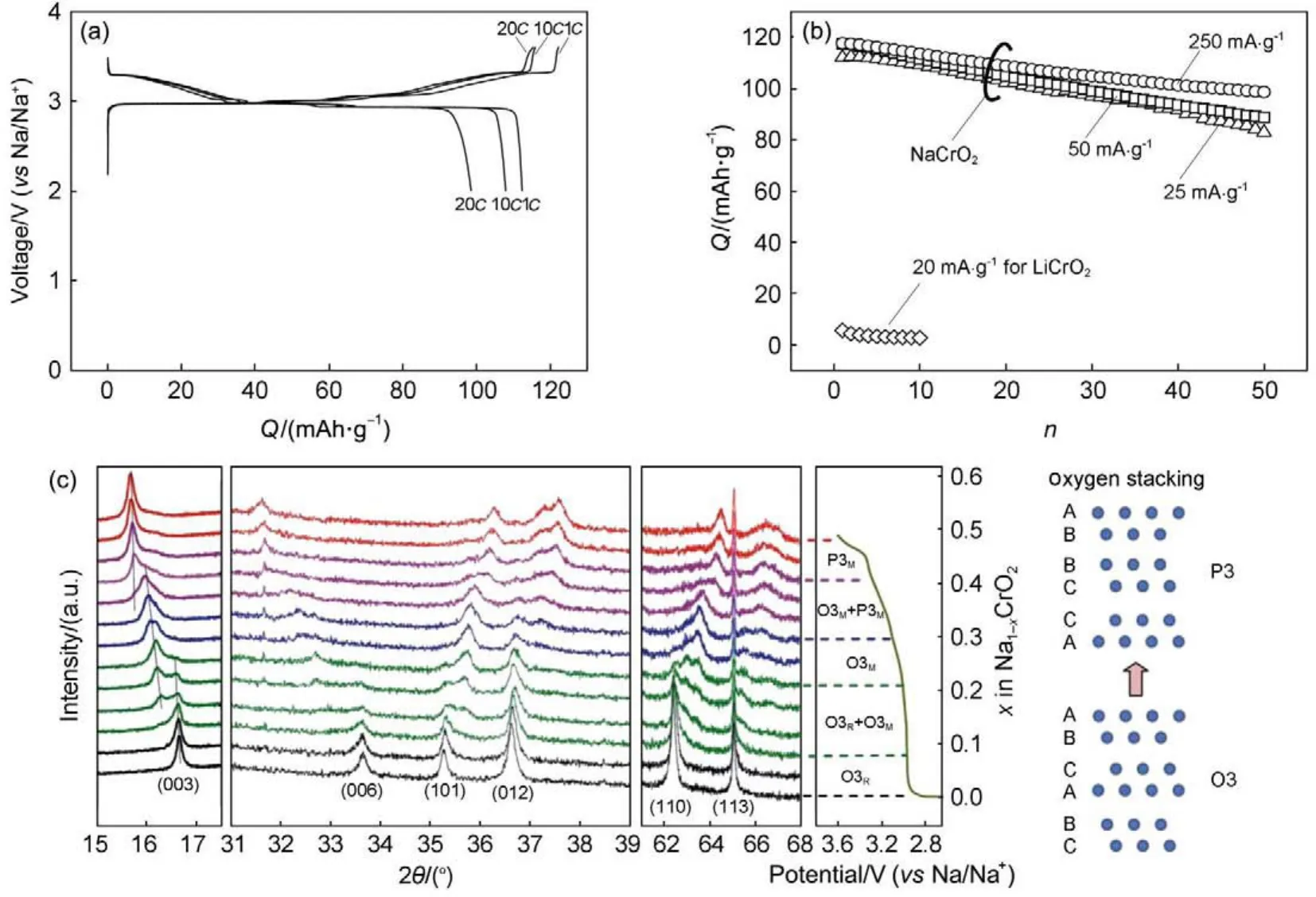
图16 (a)NaCrO2的充放电曲线、(b)NaCrO2的循环性能和(c)现场XRD59,62(a)Fig.16 (a)Charge/discharge curves and(b)cycling performance of NaCrO2,(c)in situ XRD test of NaCrO259,62(a)
O3-NaxCrO2的储钠反应在1982年就被研究,0.15Na可以可逆的嵌入脱出,对应着O3-P3相转变6。Komaba课题组59重新研究了NaxCrO2的储钠行为,该材料具有3 V左右的电压和120 mAh·g-1(0.48Na)的可逆容量,而LiCrO2电化学活性较低(图16)。同时,NaxCrO2在不同的电流密度下都表现出较好的循环稳定性。但是当充电至高电压时,同样会发生Cr4+从过渡金属层到钠层的迁移,导致容量的衰减。Ding等60通过碳包覆的方法提高了NaCrO2的电化学性能,经过碳包覆,材料具有116 mAh·g-1的比容量,循环40周,容量仍保持在110 mAh·g-1。离子液体电解液也被用于NaxCrO2电化学性能的研究61。Chen等61(b)报道了NaxCrO2在离子液体电解液中的电化学性能,该材料表现出113 mAh·g-1的可逆容量,同时具有99.6%的库伦效率,循环100周容量保持率为98%,表现出极好的稳定性。同时,非现场XRD也确认反应过程中存在着O3-Oʹ3-Pʹ3的相转变。NaxCrO2充电过程中的机理也被广泛研究62。Zhou等62(a)通过现场的同步辐射XRD和非现场的XAS研究了NaCrO2充电过程的机制,发现充电过程中存在着两个两相反应区和三个固溶体区(O3R→O3R+O3M→O3M→O3M+ P3M→P3M),晶格常数c随着充电过程而增加,a和b却减小(图16(c))。XAS表明充电到3.6 V时,Cr3+被氧化到Cr3.5+,铬离子充电过程中一直处于八面体位置,钠离子从八面体配位转为准四面体配位,最后变为棱柱配位。最近,Yu等63合成了碳包覆的O3-NaCrO2,具有优异的循环稳定性和150C的超高倍率性能。Hu等64通过选择掺入离子半径相近的Ti4+,得到P2-Na0.6Cr0.6Ti0.4O2化合物,可以有效避免Na+/空穴有序性,从而表现出较好倍率性能和循环稳定性。同时该材料由于具有不同的氧化还原中心(Cr3+,Ti4+),既可以作正极材料也可以作负极材料使用。
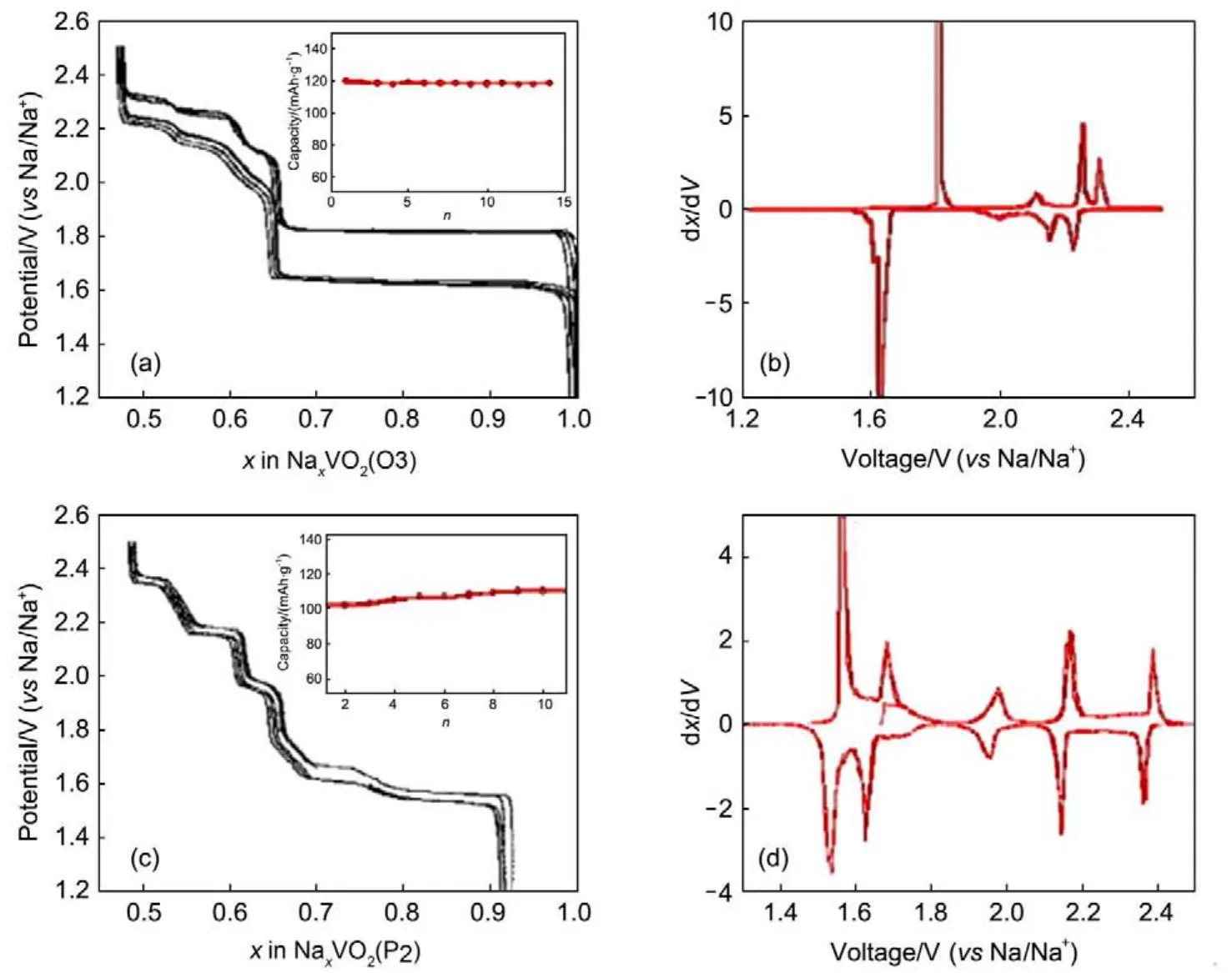
图17 NaVO2和Na0.7VO2的充放电曲线和对应的微分曲线66Fig.17 Charge-discharge curves and corresponding derivative curves of NaVO2(O3)and Na0.7VO2(P2)66
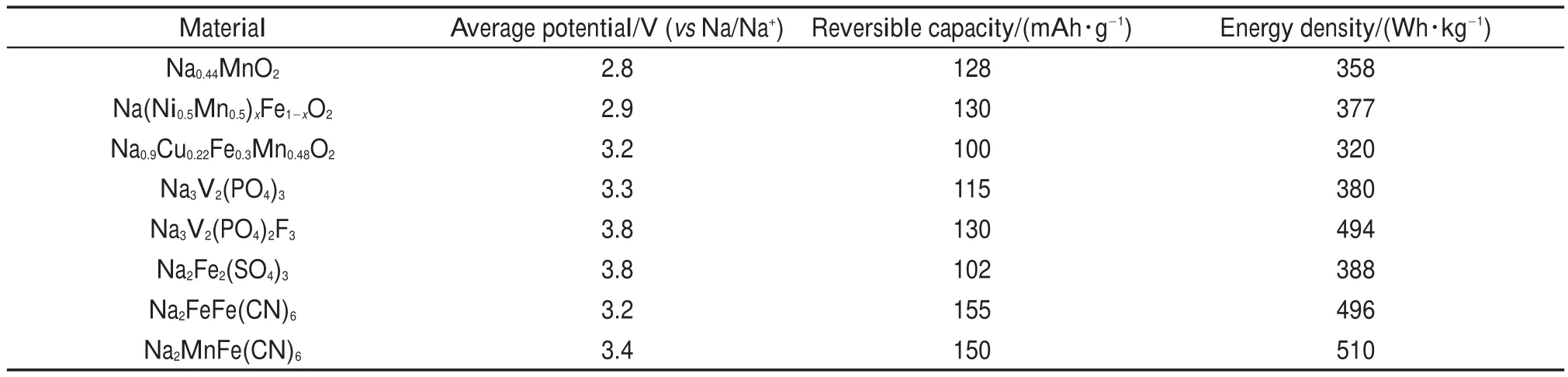
O3-NaVO2和P2-Na0.7VO2的结构和物理性质也被较早的研究和确定65。Didier等66研究了NaVO2的嵌脱钠行为,发现至少有0.5Na能够可逆脱嵌,对应着126.4 mAh·g-1的可逆容量。Hamani等67在同一时期也报道了O3-NaVO2和P2-Na0.7VO2的储钠行为,P2-Na0.7VO2比O3-NaVO2具有更小的极化,微分曲线表明两种材料具有不同的嵌脱钠机制(图17)。同时,现场XRD表明,对应P2-Na0.7VO2电极,整个过程中都表现为P2相,只是晶格常数发生一定的变化。Delmas课题组66对P2-NaxVO2和O3-NaVO2充放电过程的结构变化进行了详细研究。对于P2-NaxVO2,在0.5 上述NaxMO2(M=Co,Cr,V等)材料虽然也展现了一定的储钠性能,但主要这些元素一方面价格较高,另一方面都具有较大的毒害作用,因此将其直接用于廉价的储钠体系可能不太合适。然而对其储钠机理的研究,可发现一些新的结构和反应原理,对其它储钠反应体系提供借鉴知识也是有意义的。 2.3 聚阴离子类材料 聚阴离子类化合物一般可以表示为AxMy[(XOm)n- ]z形式,其中A为Li或Na;M为可变价态的金属离子;X为P、S、V、Si等元素。从结构上看,以X多面体与M多面体通过共边或共点连接而形成多面体框架,而A离子分布于网络的间隙中68。这类化合物作为正极材料具有如下的特点:(1)框架十分稳固,可以获得更高的循环性与安全性;(2)一些X多面体对电化学活性的Mn+/ M(n-1)+可能产生诱导效应,提升充放电的电压;(3)通过离子取代或掺杂可以调节脱嵌钠的电化学性能。但是,这类化合物存在着电子电导率和体积能量密度低的问题。聚阴离子化合物作为储钠电极材料得到广泛地研究,其中磷酸盐、焦磷酸盐和硫酸盐的电化学性能比较突出。 2.3.1 磷酸盐类化合物 磷酸盐类化合物正极材料主要包括:橄榄石型、钠快离子型导体(NASICON)、混合阴离子结构和焦磷酸盐类化合物等。 2.3.1.1 橄榄石型NaFePO4 作为LiFePO4的类似物,NaFePO4被较早的应用于钠离子电池的研究。NaFePO4有磷铁钠矿(maricite)和橄榄石(olivine)两种晶型,maricite型NaFePO4能通过高温煅烧直接合成,但是电化学活性较差;相反地,橄榄石磷酸铁钠具有较好的电化学性能,但是只能通过化学或者电化学转换橄榄石LiFePO4的方式合成69。Le Poul等70首先报道了在有机溶剂中将橄榄石LiFePO4通过化学氧化再还原的方式,转化成橄榄石型的NaFePO4。Oh等69(c)报道了通过电化学氧化再还原得到的橄榄石型NaFePO4在0.05C倍率下具有125 mAh·g-1的比容量;Zhu等71比较了NaFePO4在钠离子电池和LiFePO4在锂离子电池中的电化学性能,得出NaFePO4低的钠离子扩散系数和高的电荷转移电阻是导致NaFePO4电化学性能受限的原因。同时,钠离子嵌入脱出过程中的反应机理也被详细地研究。与LiFePO4两相反应不同的是,研究发现NaFePO4在电化学反应过程中存在一个Na2/3FePO4的中间相,即首先通过固溶体过程形成Na2/3FePO4,然后通过两相反应得到橄榄石FePO4。这样在充电曲线上表现为两个平台,如图18所示72。Lu等73通过XRD、穆斯堡尔谱以及理论计算研究了NaxFePO4的组成-温度相图,得出在2/3 考虑到LiFePO4在水溶液中稳定的电化学性能,以及水溶液电解液相对于有机电解液具有成本低、环境友好和处理简单等优势,Fang等75选用水溶液电化学转化的方法,通过LiFePO4在Li2SO4水溶液中充电,得到橄榄石型FePO4,然后,将橄榄石型FePO4在Na2SO4水溶液中放电,得到橄榄石型NaFePO4,实现了橄榄石型NaFePO4的简单快速合成。通过将转化的极片组装成钠离子电池,发现NaFePO4材料可以给出111 mAh·g-1的比容量;在0.1C倍率下循环240周,容量保持率为90%,且具有较好的倍率性能(图19)。同时,首次发现在较快的倍率下,放电曲线也能明显观察到中间相(Na2/3FePO4)的出现,并采用高分辨TEM和传统电化学方法揭示了钠离子嵌入脱出反应中的相变,计算了相转变时的反应速率常数,较好地解释了一般情况下放电曲线未出现中间相的原因。 2.3.1.2 NASICON型Na3V2(PO4)3 钠快离子导体(NASICON)具有三维的(3D)框架结构,由XO4四面体与MO6八面体构成框架(图20),钠离子位于框架形成的空隙中,具有快速的钠离子迁移率。在具有钠快离子导体结构的正极材料中,Na3V2(PO4)3为主要代表。对于Na3V2(PO4)3,钠原子处于不同的两个位点,Na1位于六配位的M1位点,Na2位于八配位的M2位点,其中M2位点的钠具有电化学活性,能够可逆地脱出和嵌入,对应的理论比容量为117 mAh·g-1,而M1位点的钠由于空间太小,无法脱出,不能提供可逆容量。 图19 水溶液电化学法合成NaFePO4和对应的NaFePO4的电化学性能75Fig.19 Aqueous electrochemical displacement method for synthesis of NaFePO4and the corresponding electrochemical performance of olivine NaFePO475 Yamaki等76首先报道了Na3V2(PO4)3在1.2-3.5 V之间有140 mAh·g-1的可逆容量。随后,Jian等77通过导电碳包覆提高材料的导电性,从而改善了材料的循环性能。其后,大量的研究工作集中于通过减小材料尺寸、表面碳包覆和元素掺杂等方式,来提升材料的电化学性能78。同时电化学反应过程中的机理也被详细研究79。Saravanan等80报道了高性能的Na3V2(PO4)3/C材料,在40C倍率下,拥有61 mAh· g-1的比容量,循环30000周,容量保持率为50%。Zhu等81报道了包埋在多孔碳中的Na3V2(PO4)3,具有200C的高倍率性能。由于Na3V2(PO4)3中的V为+3价,也可以通过先放电实现V3+/V2+的氧化还原,提供1.7 V的电压平台和50 mAh·g-1的比容量。因此,可以将Na3V2(PO4)3分别作为正、负极材料,组装成对称的钠离子全电池78(c),82。 为了进一步提高Na3V2(PO4)3材料的倍率性能和循环性能,以及寻找适合大规模应用的材料合成方法,Fang等83通过高能球磨预还原合成了Na3V2(PO4)3材料,再应用CVD技术实现原位生长出分级的高导电碳修饰的Na3V2(PO4)3/C材料,得到的Na3V2(PO4)3纳米颗粒表面具有高度石墨化的碳包覆,同时,Na3V2(PO4)3/C颗粒之间通过导电碳纤维连接,极大地提高了材料的导电性(图21)。该材料在500C的电流密度下,可逆比容量仍然可以达到38 mAh·g-1,在30C倍率下循环20000周,容量保持率为54%,是目前报道的性能最优异的Na3V2(PO4)3/C材料。同时,他们以该Na3V2(PO4)3/C材料为正极,以石墨烯修饰的NaTi2(PO4)3材料为负极,构造了全NASICON的全电池,全电池具有128 mAh·g-1的可逆比容量,并具有较好的功率性能和循环稳定性84。选择构建分级的高导电性网络,对于获得高性能电极材料,提供了参考和借鉴。 图20 Na3V2(PO4)3的结构示意图Fig.20 Structure of Na3V2(PO4)3 通过元素掺杂,也可以提高材料的电化学性能。Mason等85通过掺入Fe原子,可以激活V4+/5+的电化学活性,得到4 V的电位平台,并且可以增加约12%的容量。Lim等86研究发现K离子掺杂可以增加晶体的体积,提高钠离子的扩散空间以及稳定结构,从而提高材料的倍率性能。Li等87通过Mg掺杂可以取代V的位点,继而提高材料的倍率性能和循环稳定性。然而一些元素掺杂的系统研究和理论分析仍然不足,对选择合适的掺杂元素和比例尚存盲目性。 图21 Na3V2(PO4)3电化学性能83Fig.21 Electrochemical performance of Na3V2(PO4)383 2.3.1.3 焦磷酸盐(Na2MP2O7) Na2MP2O7(M=Fe,Mn)属于三斜晶系,P1空间群,以FeO6八面体和PO4四面体,通过共顶点氧原子,产生[011]方向的钠离子扩散通道,因此表现出储钠电化学活性。结构中也存在着P2O7的单元,以共角或共边的形式形成Fe2O11二聚体。焦磷酸盐以Na2FeP2O7性能最为优异,研究比较广泛。Barpanda等88首先通过固相法合成了Na2FeP2O7,并测试了该材料的储钠性能,该材料平均电压在3 V,并表现出82 mAh·g-1的可逆比容量(图22)。他们接着研究了脱钠后NaFeP2O7的热稳定性89,发现该材料在560°C会由P1相转为P21/C相,但是不会发生分解,这种稳定性得益于(P2O7)4-高的热稳定性。Kim等90通过准平衡反应测试和第一性原理分析了Na2FeP2O7储钠反应的机理,得出NaFeP2O7在2.0-4.5 V之间存在两种反应:2.5 V左右的两相反应和3.0-3.25 V之间一系列的两相反应。同时,Park和Barpanda几乎同时报道了三斜晶系Na2MnP2O7的储钠性能91,如图22(c)所示,这一材料可以输出3.6 V的电压和90 mAh·g-1的放电比容量,对应于Mn3+/Mn2+的氧化还原。同时,Na2Fe1-xMnxP2O7的材料也被报道,但Mn的掺入会降低材料的容量92。另外,Barpanda等93也报道了Na2CoP2O7具有3 V的平均电压和80 mAh·g-1的容量,表现出倾斜的充放电曲线。而他们报道的t-Na2(VO)P2O7也具有3.8 V的电压和80 mAh·g-1的容量94。最近,Kim等95报道了一种新的电极材料Na7V3(P2O7)4,具有4.13 V的电压平台和80 mAh· g-1的可逆比容量,同时,在1C倍率下循环600周,容量保持率为75%,表现出较好的循环稳定性。 图2 2Na2FeP2O7的晶体结构图(a)以及Na2FeP2O7(b)和Na2MnP2O7(c)的充放电曲线88,91(a)Fig.22 Structure of Na2FeP2O7(a)and charge/discharge profiles of Na2FeP2O7(b)and Na2MnP2O7(c)88,91(a) 由焦磷酸盐的结构和性能可知,焦磷酸盐可以直接通过高温煅烧得到电化学活性较好的材料,针对铁基化合物具有较好的优势。但是,由于含有较大的P2O7离子,导致材料的分子量较大,理论容量偏低。 图2 3Na2Fe2(SO4)3的晶体结构图和充放电曲线96Fig.23 Crystal structure and chare/discharge proffiles of Na2Fe2(SO4)396 2.3.2 硫酸盐 硫酸盐的储钠性能也被广泛地研究,其中Fe基硫酸盐表现出最为优异的电化学性能。Barpanda等96制备了alluaudite型的Na2Fe2(SO4)3,该材料表现出3.8 V的电压,并且具有102 mAh·g-1的可逆比容量,在20C的倍率下仍有55 mAh·g-1的理论比容量(图23)。同时,理论计算以及穆斯堡尔谱分析该材料存在一些杂相。Yamada等97分析了非整比的Na2+2xFe2-x(SO4)3,发现x=0.25的时候,杂相含量最小,由此Na2.4Fe1.8(SO4)3的结构被广泛地研究。Meng等98通过自上而下的方法合成了单壁碳纳米管修饰的Na2+2xFe2-x(SO4)3材料,该材料表现出优异的倍率性能和循环稳定性。Wei等99研究了Mn掺杂的Na2.5(Fe1-yMny)1.75(SO4)3(0≤y≤1)的电化学性能,发现随着Mn的掺入,Fe3+/2+氧化还原电位会升高,但是材料的容量会降低。Goodenough等100报道了Eldfellite相的NaFe(SO4)2,该材料表现出3V的电压平台,拥有99 mAh·g-1的可逆比容量。同时,类似的Na2+2xMn2-x(SO4)3、Na2Mg(SO4)2·4H2O等也被研究,但性能较差101。 目前研究的硫酸盐体系,Na2.4Fe1.8(SO4)3材料的综合性能最优,其具有较高的工作电压以及合适的容量,加之铁基材料价格低廉、环境适应性好,通过寻找适合大规模合成的方法,构造合适的导电网络,有可能获得较好的电化学性能,具有潜在的应用前景。 2.3.3 混合阴离子化合物 对于聚阴离子化合物,通过使用混合的阴离子,可以构造出新的结构体系,获得较好的电化学活性;同时,选择强的吸电子基团(如F等),可以通过诱导作用提高材料的电压。混合的聚阴离子体系由Goodenough等102较先在锂离子电池电极材料进行研究,指的是以两种不同的阴离子组成的化合物,具有稳定电化学性能的主要是XO4F (X=P,S)和PO4P2O7等。 2.3.3.1 XO4F 通过引入强电负性的F,可以提高材料的工作电压,从而提高材料的能量密度。以PO4F组成的化合物研究比较多的主要是Na2FePO4F和Na3V2(PO4)2F3。 Nazar等103报道了Na2FePO4F的合成,该材料可同时应用于钠离子电池和锂离子电池。Tarascon等104比较了固相法和离子热法合成的Na2FePO4F的电化学性能,发现通过离子热法合成的材料颗粒较小,电化学性能较好。他们同时研究了Mn掺杂后的Na2Fe1-xMnxPO4F的电化学性能。随后,Komaba等105(a)通过碳包覆提高Na2FePO4F的导电性,从而极大的提高了材料的电化学性能(图24)。这些材料的平均电压都要高于相应非氟取代的材料体系,因此,通过选择氟取代,可以获得较高工作电压的电极材料,这对寻找新的电极材料提供了借鉴。 图24 Na2FePO4F的电化学性能105(a)Fig.24 Electrochemical performances of Na2FePO4F105(a) Le Meins等106首先报道了Na3V2(PO4)2F3的晶体结构,随后,Na3V2(PO4)2F3被作为锂离子电池电极材料得到广泛研究107。Kang等108第一次通过第一性原理结合实验的方法研究了Na3V2(PO4)2F3的储钠机制,确定储钠过程为单相反应。这种材料的电压平台为3.7和4.2 V,说明F取代可以提高充放电电压。该材料表现出108 mAh·g-1的可逆容量,循环30周容量几乎保持不变(图25(a))。随后,Na3V2(PO4)2F3的嵌脱钠机制和结构演化得以更详细地研究109。 以O部分替代F可以制备出一类具有新型结构的化合物Na3V2O2x(PO4)2F3-2x,Massa等110第一次合成并解析了Na3V2O2(PO4)2F的结构。Sauvage等111随后将该材料应用于钠电池,该材料在3.6和4.0V处出现两个放电平台,给出87 mAh·g-1的比容量(图25(b))。Serras等112研究了不同的碳类型以及碳包覆量对Na3V2O2x(PO4)2F3-2x电化学性能的影响,可以得到120 mAh·g-1的比容量。Peng等113研究了RuO2包覆的Na3V2O2(PO4)2F的纳米线,具有优异的倍率性能和循环稳定性。Qi等114通过溶剂热法合成了一系列Na3V2O2x(PO4)2F3-2x纳米材料,其中,Na3(VOPO4)2F材料具有10C的倍率性能,并能稳定循环1200周。随后,其电化学性能和反应机理得到广泛而深入地研究115。 同时,一些F掺杂的硫酸盐材料也有相关报道,如NaFeSO4F,但它们的储钠性能较差,研究也比较少116。 2.3.3.2 (PO4)(P2O7) Kim等117通过固相法合成了Na4Fe3(PO4)2(P2O7)材料,并测试了其电化学性能,该材料平均电压为3.2 V,具有129 mAh·g-1的可逆比容量(图26 (a))。随后,他们又对Na4Fe3(PO4)2(P2O7)材料储钠反应的机理进行了研究,确定嵌脱钠过程为单相变化,并且体积变化只有4%118。 图25 Na3V2(PO4)2F3和Na3V2O2x(PO4)2F3-2x的充放电曲线109b,110Fig.25 The electrochemical performance of Na3V2(PO4)2F3and Na3V2O2x(PO4)2F3-2x109b,110 Nose等119研究了含Co的Na4Co3(PO4)2(P2O7)材料储钠性能,该材料在4.1-4.3 V有一系列氧化还原电对,并具有95 mAh·g-1的可逆比容量和较好的倍率性能。他们继续研究掺入Mn和Ni后得到的Na4Co2.4Mn0.3Ni0.3(PO4)2P2O7材料的电化学性能120,Mn、Ni、Co都有电化学活性,充放电曲线变得平滑。Wood等121随后对这类材料Na4M3(PO4)2(P2O7) (M=Fe,Mn,Co,Ni)进行了系统地理论分析,如原子能最小化计算、分子动力学计算、密度泛函理论计算等,分析了材料结构缺陷、钠离子扩散路径、电压变化等。Deng等122合成出了一种新的化合物Na7V4(P2O7)4(PO4),具有3.8 V的平台,表现出92 mAh·g-1的可逆比容量(图26(c))。 通过磷酸根和焦磷酸根的复合,可以提高材料的电压,以及探索出新的结构体系,但是较大的阴离子基团,导致材料具有低的理论容量,需要探索一些新的结构体系。 2.3.3.3 CO3PO4 聚阴离子化合物具有稳定的框架结构以及较大的钠离子传输间隙,具有长的循环稳定性和安全性,提高材料的电子电导率,可以获得优异的电化学性能,具有潜在的应用前景。通过选择不同的阴离子基团,构造出混合阴离子体系,可以获得新的结构和新的材料体系。在聚阴离子类材料中,Na3V2(PO4)3、Na3V2(PO4)2F3、Na2FePO4F、Na4Fe3(PO4)2(P2O7)等由于具有较大的离子隧道和优异的电化学稳定性,将具有潜在的应用前景。 2.4 普鲁士蓝类大框架化合物 普鲁士蓝类化合物的常见组成为AxMA[MB(CN)6]·zH2O(A为碱金属离子,MA和MB为过渡金属离子),其结构为面心立方结构,如图28所示。过渡金属离子分别与氰根中的C和N形成六配位,碱金属离子处于三维通道结构和配位孔隙中。这种大的三维多通道结构可以实现碱金属离子的嵌入和脱出。同时,通过选用不同的过渡金属离子,如Ni2+、Cu2+、Fe2+、Mn2+、Co2+等,可以获得丰富的结构体系,表现出不同的储钠性能。 2.4.1 AxFeFe(CN)6 铁基材料由于资源丰富、成本低廉以及结构稳定等特点,得到广泛地关注。Qian等126首先报道了Na4Fe(CN)6在钠离子电池中的应用,材料具有87 mAh·g-1比容量,并表现出非常优异的循环性能(图28b)。随后,他们通过快速沉淀法制备了NaFeFe(CN)6材料,材料首周充放电比容量为95和113 mAh·g-1,并表现出较好的循环稳定性127。Goodenough等128报道的KFeFe(CN)6材料表现出100 mAh·g-1的可逆比容量,且循环30周容量保持不变。 图26 几类Na4M3(P2O7)4(PO4)的充放电曲线118,119,122(b)Fig.26 Charge-discharge curves of Na4M3(P2O7)4(PO4)118,119,122(b) Wu等129通过缓慢结晶的方法,合成出低缺陷的FeFe(CN)6化合物,由于具有较少的空位和晶格水分子,该材料的容量和循环性能得到极大的提升,材料具有120 mAh·g-1的可逆容量,循环500周容量保持率为87%,但是贫钠态限制了材料的实际应用。Huang等130通过共沉淀法合成了Na/Fe=0.85的化合物,该材料首周充放电比容量为94和120 mAh·g-1,循环100周容量保持率为76%。Guo等131合成了低缺陷和低含水量的Na0.61Fe [Fe(CN)6]0.94,可以实现两个Na+的嵌入脱出(170 mAh·g-1),循环150周,容量几乎不变。为了合成完全富钠态的普鲁士蓝Na2FeFe(CN)6,他们通过在氮气保护和添加还原剂(Vc)防止Fe2+氧化的条件下,制备出Na1.61Fe1.89(CN)6材料,获得150 mAh·g-1的首周充电容量,同时具有较好的循环稳定性132。Goodenough等128通过水热法成功合成了Na1.92FeFe (CN)6材料,可以给出155 mAh·g-1的可逆比容量(图28(c)),循环750周,容量保持率为80%,同时,与硬碳组成全电池表现出较好的电化学性能133。 图27 两类Na3MPO4CO3的充放电曲线124,125Fig.27 Charge-discharge curves of Na3MPO4CO3124,125 2.4.2 AxMnFe(CN)6 锰由于价格低廉,同样受到关注。Goodenough等134首先报道了Na2MnFe(CN)6材料的储钠性能,较高Na含量的Na1.72Mn[Fe(CN)6]0.99材料具有3.3 V的电压和40C的倍率性能。随后,他们通过除尽晶格中的结晶水,得到一种扭曲的晶体结构,合成的Na2MnFe(CN)6具有极为平坦的充放电曲线,循环500周容量保持率为75%,并表现出非常好的循环稳定性135(图29)。Ma等136通过掺入12%的Ni,可以极大的提高材料的循环稳定性,材料具有118 mAh·g-1的可逆容量;同时,循环800周容量保持率为84%。Chou等137通过PPy包覆Na2MnFe(CN)6材料,不仅提高了材料的电子电导率,同时可以抑制锰离子的流失而提高材料的稳定性,另外,还可以抑制晶格中水分子在高电位下的氧化分解,聚合物可以进行p掺杂来提高复合材料的容量。 图28 普鲁士蓝结构图和充放电曲线126,133Fig.28 Structure and charge-discharge curves of ferrocyanides126,133 2.4.3 AxCoFe(CN)6 钴类材料由于具有较高的氧化还原电对以及两电子的反应,也引起了学者的关注。Kang等138较早通过软X射线吸收谱研究了NaCo[Fe(CN)6] H2O纳米颗粒的电子结构,发现铁和钴都分别表现出Fe2+-Fe3+(2.36价)和Co2+-Co3+(2.41价)的混合价态,并且Fe和Co对O和CN分别表现出不同的配位能力。Tomoyuki等139通过电沉积的方法制备了Na1.6Co[Fe(CN)6]0.9·2.9H2O薄膜,可以获得135 mAh·g-1的可逆容量以及60C的倍率性能,该材料在3.8和3.4 V处拥有两个放电平台,对应着Fe2+/ Fe3+和Co2+/Co3+的氧化还原,同时,他们通过非现场XRD研究了电化学反应过程中的结构变化。Wu等140通过掺入适量的Ni提高普鲁士蓝结构的稳定性,得到的Na2Ni0.4Co0.6Fe(CN)6具有90 mAh·g-1的比容量,并且在800 mA·g-1的电流密度下具有69 mAh·g-1的可逆容量。最近,Yang等141通过控制结晶的方法,合成了低缺陷高结晶性的Na2CoFe(CN)6材料,该材料具有150 mAh·g-1的可逆容量,同时循环200周容量保持率为90%,表现出较好的电化学稳定性(图30)。 2.4.4 AxMFe(CN)6(M=Ni,Cu) 含Ni、Cu的普鲁士蓝类化合物在水溶液电池中研究比较广泛,有机钠离子电池体系研究较少。Guo等142研究了KNiFe(CN)6在有机电解液中的储钠性能,该材料具有66 mAh·g-1的可逆容量,同时能够稳定循环200周。Dai等143合成了介孔的Na2NiFe(CN)6,该材料具有65 mAh·g-1的可逆容量,同时,循环180周,容量几乎无衰减。 Talham等144合成了K0.1Ni[Fe(CN)6]0.7·4.4H2O包覆的K0.1Cu[Fe(CN)6]0.7·3.5H2O材料,复合物材料具有75 mAh·g-1的可逆容量和3.3V左右的平台,在600 mA·g-1的电流密度下具有约20 mAh·g-1的可逆容量。 普鲁士蓝类化合物具有较高的电压和可逆容量,并且成本较低,具有潜在的应用前景。但是,循环稳定性有待改善,材料极易形成缺陷,影响材料整体的容量和电化学性能,且材料高温受热易分解,存在一定的安全隐患。 2.5 有机化合物和聚合物 有机物正极材料具有理论比容量高、原料丰富、环境友好、价格低廉和结构设计灵活的优点,是一类具有广泛应用前景的储能物质。正极材料要求具有较高的氧化还原电势,研究较多的正极材料主要包括含醌、酸酐、酰胺以及酚类等的有机小分子,以及聚合物。 2.5.1 有机小分子正极 作为钠离子电池正极材料,有机化合物相比于无机材料研究较少。小分子有机化合物得到广泛的研究。Chen等145将蒽醌分子与多孔碳CMK-3复合,得到的材料首周具有214 mAh·g-1的可逆容量,循环50周,容量保持率为88%。Ji等146研究了苝四羧酸酐染料的储钠性能,该材料具有2.3 V左右的电压平台和145 mAh·g-1的可逆容量,同时,在1000 mA·g-1的电流密度下,仍有91 mAh·g-1的可逆容量,表现出较好的倍率性能和循环稳定性。Deng等147研究了苝四酰亚胺分子的储钠行为,该材料具有大的共轭苝环结构,难溶于电解液,材料具有2 V左右的电压和140 mAh·g-1的可逆容量,循环300周,容量保持率为90%。Yao等148报道了有机染料靛蓝作为电极材料,该材料具有1.8 V左右的电压和100 mAh·g-1的可逆容量。为了获得更高的容量,Chihara等149报道了一类Na2C6O6,该材料可以实现两个钠离子的嵌入脱出,给出270 mAh·g-1的可逆比容量。Chen等150报道了一类双极性有机小分子,该材料为含有羧基基团和烯醇式结构的单苯四钠盐Na4DHTPA,材料可以同时作为正极和负极,并分别可以给出大于180 mAh·g-1的可逆容量,且循环性能良好。同时,他们将该材料作为正负极,构造全有机材料的全电池,全电池具有1.8 V的电压和65 Wh·kg-1的能量密度(图31)。 图29 Na2-δMnFe(CN)6的结构和充放电曲线135Fig.29 Structure and charge/discharge profiles of Na2-δMnFe(CN)6135 图30 Na2CoFe(CN)6的电化学性能141Fig.30 Electrochemical performance of Na2CoFe(CN)6141 2.5.2 聚合物正极 小分子电极材料由于在电解液中溶解度较大,电化学性能受到一定的限制。有机聚合物具有很长的链段结构,难溶于有机电解液,具有更好的稳定性。Zhao等151合成了苯胺-硝基苯胺共聚物,该材料具有3.2 V的放电电压,首周可逆容量可以达到180 mAh·g-1,循环50周仍有173 mAh· g-1的容量,如此优异的电化学性能证明聚合物确实具有有效的储钠性能(图32a)。随后,Zhou等152率先通过向电活性的聚合物中掺入不溶的和氧化还原活性的铁氰根阴离子,直接改变了导电聚合物的反应机制,从传统的p掺杂/脱杂转为阳离子的嵌入脱出、的氧化还原和阴离子掺杂的协同进行,使得聚合物的电化学活性得到极大的应用,也提高了材料的循环稳定性。通过掺入氧化还原活性的可以得到135 mAh·g-1的可逆比容量和较好的循环稳定性,循环100周容量保持率为85%(图32(b))。同时,Zhou153又继续研究了二苯胺磺酸根掺杂的聚吡咯的电化学性能,该材料可以给出115 mAh·g-1的可逆比容量,以及较好的循环稳定性和倍率性能。 图31 以Na4DHTPA为正负极的全有机电池的电化学性能150Fig.31 Electrochemical performance offull cell constructed of Na4DHTPA150 图32 几类聚合物的充放电曲线151,152,154Fig.32 Charge-discharge curves of some polymers151,152,154 为了实现有效的储钠反应,Zhu等154首次合成了自掺杂的聚合物,带有接枝的烷基磺酸钠,从而从根本上改变聚合物p掺杂脱杂的机理,实现完全的钠离子嵌入脱出反应。该有机化合物具有3 V左右的放电平台,同时可以给出85 mAh·g-1的可逆比容量,并能稳定循环100周(图32(c))。Zhang等155选用不同的二酸酐和二胺单体,合成了一系列聚酰亚胺化合物,并测试了他们的电化学性能,其中,基于二羧酸酐PTCDA的聚酰亚胺具有2.2 V最高的氧化还原电位,且循环5000周容量保持率为98%,表现出非常好的循环稳定性。Xu等156合成了含亚胺和蒽醌氧化还原活性基团的聚合物PI1和PI2,两种聚酰亚胺化合物具有2 V左右的电压和200 mAh·g-1比容量,循环200周容量保持率高达90%以上。为了提高聚酰亚胺类材料的氧化还原电位,Xu等157选用具有强吸电子性、低分子量的单体合成了聚磺酰亚胺化合物,材料具有2.3 V的电压和150 mAh·g-1的可逆容量。为了探索更高电压的材料,Xie等158最早研究了一种自由基聚合物正极(聚降冰片烯衍生物)的储钠行为,该化合物可以通过氮氧自由基的氧化,同时电解液中的阴离子嵌入(p掺杂),实现每个单元一个电子的转移。他们研究的这种化合物具有3.38 V的电压平台和75 mAh·g-1的可逆比容量,但循环50周,容量保持率只有64.5%。 Deng等159合成了聚三苯胺化合物,同时与聚硫化蒽醌负极组成全有机电池,该全电池具有1.8 V的电压和92 Wh·kg-1的能量密度,同时循环500周,容量保持率为85%,为全有机钠离子电池的应用提供了可能。最近,Banda等160也研究了聚酰亚胺类有机物的储钠性能,并组装成全有机钠离子电池,具有1.35 V的放电电压。 有机电极材料不含过渡金属元素,环境友好、价格低廉、种类多样,并且可以根据结构合理设计,化合物灵活多变,具有广阔的前景。构造合适的结构,提高有机材料的电压与循环稳定性,减少材料在电解液中的溶解度,将具有重要意义。 图33 非晶介孔FePO4的电化学性能164Fig.33 Electrochemical performance of mesoporous amorphous FePO4164 2.6 非晶化合物 非晶化合物,又称为无定型化合物,是固体中的原子不按照一定的空间顺序排列的固体,原子排布上表现为长程无序而短程有序。非晶化合物由于没有晶格限制,钠离子在颗粒表面反应,不会引起材料结构的变化,因而可以表现出更好的稳定性。负极材料比较容易形成非晶相,如碳材料、磷、二氧化钛等,而正极材料较难形成非晶相。 FePO4很容易形成非晶相,其作为钠离子电池研究比较广泛。Shiratsuchi等161首先报道了无定型FePO4作为钠离子电池电极的储钠行为,在0.1 mA·cm-2的电流密度下,有146 mAh·g-1的可逆比容量,表现出与锂离子电池相似的性能,表明钠离子与锂离子具有相似的反应位点。Zhao等162通过溶剂萃取的方法,合成了单分散的FePO4·2H2O纳米球,具有108 mAh·g-1的容量。Liu等163报道了单壁碳纳米管修饰的FePO4纳米颗粒,具有120 mAh·g-1的可逆比容量。这些研究报道的FePO4的可逆容量都远低于它的理论比容量(178 mAh· g-1),循环性能也有待改善。Fang等164通过化学诱导沉淀的方法,合成了介孔的非晶FePO4,通过与导电碳球磨提高材料的导电性,所得到的材料具有151 mAh·g-1的可逆比容量,循环160周容量保持率为94%,具有极好的循环稳定性(图33)。Liu等165合成了玉米棒型的FePO4多壁碳纳米管复合物,具有155 mAh·g-1的可逆比容量,循环70周容量几乎不变。Wang等166对比了晶型和非晶的FePO4的储钠性能,发现非晶FePO4可以表现出比晶型更优的电化学性能。FePO4由于贫钠,限制了它的应用,Li等167直接合成了无定型的NaFePO4纳米球,具有154 mAh·g-1的可逆比容量,循环300周,容量保持率为95%,表现出极好的稳定性,同时,该材料还具有10C的倍率性能。 Uchaker等168也对比了非晶和晶型的V2O5材料的储钠性能,发现非晶V2O5可以给出241 mAh·g-1的可逆比容量,相当于两倍的结晶的V2O5的可逆容量,同时,非晶V2O5也表现出更高的放电电压、能量密度和循环稳定性。Fu等169合成了介孔的非晶FeOF,可以给出239 mAh·g-1的可逆比容量,以及优异的循环稳定性和倍率性能。 表1 几种钠离子电池正极材料电化学性能比较Table 1 Comparisons of the electrochemical performance of several cathode materials for SIBs 非晶材料没有晶格的限制,构造合适的非晶材料,有可能获得优于结晶材料的电化学性能。同时,对非晶材料的反应机理的研究将具有重要意义。 受环境污染的加重和锂资源价格不断上涨的影响,资源丰富、价格低廉、环境友好的钠离子电池将越来越受到大家的重视。钠离子电池正极材料是提高钠离子电池性能的关键材料之一,目前钠离子电池正极材料取得了一些研究成果(见图2)。层状氧化物具有较高的容量和充放电电压,但由于充放电过程中存在着许多相变,长期的循环将导致结构的坍塌,而通过元素取代或掺杂可以在一定程度上平滑充放电曲线,抑制结构相变,提高循环稳定性;聚阴离子化合物由于具有稳定的框架结构,因此具有稳定的电化学性能和较高的热安全性,但是较大的阴离子基团导致聚阴离子化合物容量普遍不高;普鲁士蓝类材料具有大的隧道结构,循环过程中结构比较稳定,但是材料的振实密度较低,结晶水难以除去,且存在受热安全隐患;非晶材料结构上没有晶格限制,钠离子在嵌入脱出过程中,对材料的结构应力较小,材料具有稳定的电化学性能,是将来发展储钠正极材料结构和体系的一个新的方向,但目前可利用的非晶富钠正极体系较少,需要在合成和结构设计上提出新的构想。从目前储钠正极材料的应用发展来看,虽然在结构类型和材料体系上已有较多工作,也展现了较好的电化学性能(容量、倍率和循环等),然而若从综合性能(包括资源利用、环境友好、容量、效率、倍率和循环等)方面来考虑,可用于实际大规模钠离子电池的正极材料体系仍不多(表1),如过渡金属氧化物应该集中在资源丰富的材料体系(Mn,Fe,Ti,Cu或少量Ni);对于聚阴离子体系,Na3V2(PO4)3和Na3V2(PO4)2F3应该适合于高能量密度和长寿命的钠离子电池体系;若解决好Na2Fe2(SO4)3的循环效率和稳定性,也可以作为廉价的储钠正极材料进行大规模应用;对于高容量的普鲁士蓝类材料(Na2FeFe(CN)6、Na2MnFe(CN)6等),若解决了循环稳定性和其受热分解所引起的安全隐患等问题,也是极具应用前景的高性能储钠正极材料。总的来说,氧化物材料由于电化学反应过程中经历较多的相变和结构变化,循环寿命受到一定的限制;聚阴离子类材料由于结构稳定,可以获得较长的循环寿命和较高的倍率,但较大的阴离子基团使得材料的能量密度受到一定的限制;普鲁士蓝类化合物具有较高的能量密度,寻找合适的合成方法来获得较高纯度的材料成为关键。总之,近五年,储钠正极材料的研究备受关注,新结构和新体系层出不穷,这势必会大大推动钠离子电池的应用发展。 (1)Tarascon,J.M.Nat.Chem.2010,2,510.doi:10.1038/ nchem.680 (2)Fang,Z.;Cao,Y.L.;Hu,Y.S.;Chen,L.Q.;Huang,X.J. Energy Storage Sci.Technol.2016,5,149.[方 铮,曹余良,胡勇胜,陈立泉,黄学杰.储能科学与技术,2016,5,149.] doi:10.3969/j.issn.2095-4239.2016.02.005 (3)Mizushima,K.;Jones,P.C.;Wiseman,P.J.;Goodenough,J. B.Mater.Res.Bull.1980,15,783.doi:10.1016/0025-5408(80) 90012-4 (4)(a)Braconnier,J.J.;Delmas,C.;Fouassier,C.;Hagenmuller, P.Mater.Res.Bull.1980,15,1797.doi:10.1016/0025-5408 (80)90199-3(b)Delmas,C.;Braconnier,J.J.;Fouassier,C.;Hagenmuller, P.Solid State Ionics 1981,3-4,165.doi:10.1016/0167-2738 (81)90076-X (5)Abraham,K.M.Solid State Ionics 1982,7,199.doi:10.1016/ 0167-2738(82)90051-0 (6)Braconnier,J.J.;Delmas,C.;Hagenmuller,P.Mater.Res.Bull. 1982,17,993.doi:10.1016/0025-5408(82)90124-6 (7)Mendiboure,A.;Delmas,C.;Hagenmuller,P.J.Solid State Chem.1985,57,323.doi:10.1016/0022-4596(85)90194-X (8)Yabuuchi,N.;Kubota,K.;Dahbi,M.;Komaba,S.Chem.Rev. 2014,114,11636.doi:10.1021/cr500192f (9)Delmas,C.;Fouassier,C.;Hagenmuller,P.Physica B+C 1980, 99,81.doi:10.1016/0378-4363(80)90214-4 (10)Blangero,M.;Carlier,D.;Pollet,M.;Darriet,J.;Delmas,C.; Doumerc,J.P.Phys.Rev.B 2008,77,184116.doi:10.1103/ PhysRevB.77.184116 (11)Doeff,M.M.;Peng,M.Y.;Ma,Y.;De Jonghe,L.C.J. Electrochem.Soc.1994,141,L145.doi:10.1149/1.2059323 (12)Sauvage,F.;Laffont,L.;Tarascon,J.M.;Baudrin,E.Inorg. Chem.2007,46,3289.doi:10.1021/ic0700250 (13)Cao,Y.;Xiao,L.;Wang,W.;Choi,D.;Nie,Z.;Yu,J.;Saraf,L. V.;Yang,Z.;Liu,J.Adv.Mater.2011,23,3155.doi:10.1002/ adma.201100904 (14)Kim,H.;Kim,D.J.;Seo,D.H.;Yeom,M.S.;Kang,K.;Kim, D.K.;Jung,Y.Chem.Mater.2012,24,1205.doi:10.1021/ cm300065y (15)(a)Hosono,E.;Saito,T.;Hoshino,J.;Okubo,M.;Saito,Y.; Nishio-Hamane,D.;Kudo,T.;Zhou,H.J.Power Sources 2012,217,43.doi:10.1016/j.jpowsour.2012.05.100 (b)Qiao,R.;Dai,K.;Mao,J.;Weng,T.C.;Sokaras,D.; Nordlund,D.;Song,X.;Battaglia,V.S.;Hussain,Z.;Liu,G.; Yang,W.Nano Energy 2015,16,186.doi:10.1016/j. nanoen.2015.06.024 (c)Wang,C.H.;Yeh,Y.W.;Wongittharom,N.;Wang,Y.C.; Tseng,C.J.;Lee,S.W.;Chang,W.S.;Chang,J.K.J.Power Sources 2015,274,1016.doi:10.1016/j.jpowsour.2014.10.143 (d)Demirel,S.;Oz,E.;Altin,E.;Altin,S.;Bayri,A.;Kaya,P.; Turan,S.;Avci,S.Mater.Charact.2015,105,104. doi:10.1016/j.matchar.2015.05.005 (16)Guo,S.;Yu,H.;Liu,D.;Tian,W.;Liu,X.;Hanada,N.;Ishida, M.;Zhou,H.Chem.Commun.2014,50,7998.doi:10.1039/ c4cc02362e (17)Jiang,X.;Liu,S.;Xu,H.;Chen,L.;Yang,J.;Qian,Y.Chem. Commun.2015,51,8480.doi:10.1039/c5cc02233a (18)(a)Wang,Y.;Liu,J.;Lee,B.;Qiao,R.;Yang,Z.;Xu,S.;Yu, X.;Gu,L.;Hu,Y.S.;Yang,W.;Kang,K.;Li,H.;Yang,X.Q.; Chen,L.;Huang,X.Nat.Commun.2015,6.doi:10.1038/ ncomms7401 (b)Wang,Y.;Mu,L.;Liu,J.;Yang,Z.;Yu,X.;Gu,L.;Hu,Y. S.;Li,H.;Yang,X.Q.;Chen,L.;Huang,X.Adv.Energy Mater.2015,doi:10.1002/aenm.201501005 (19)Xu,S.;Wang,Y.;Ben,L.;Lyu,Y.;Song,N.;Yang,Z.;Li,Y.; Mu,L.;Yang,H.T.;Gu,L.;Hu,Y.S.;Li,H.;Cheng,Z.H.; Chen,L.;Huang,X.Adv.Energy Mater.2015,doi:10.1002/ aenm.201501156 (20)Ma,X.;Chen,H.;Ceder,G.J.Electrochem.Soc.2011,158, A1307.doi:10.1149/2.035112jes (21)Abakumov,A.M.;Tsirlin,A.A.;Bakaimi,I.;Van Tendeloo, G.;Lappas,A.Chem.Mater.2014,26,3306.doi:10.1021/ cm5011696 (22)Caballero,A.;Hernan,L.;Morales,J.;Sanchez,L.;Santos Pena,J.;Aranda,M.A.G.J.Mater.Chem.2002,12,1142. doi:10.1039/b108830k (23)Paulsen,J.M.;Dahn,J.R.Solid State Ionics 1999,126,3. doi:10.1016/S0167-2738(99)00147-2 (24)Yuan,D.;He,W.;Pei,F.;Wu,F.;Wu,Y.;Qian,J.;Cao,Y.;Ai, X.;Yang,H.J.Mater.Chem.A 2013,1,3895.doi:10.1039/ c3ta01430d (25)Yabuuchi,N.;Hara,R.;Kajiyama,M.;Kubota,K.;Ishigaki, T.;Hoshikawa,A.;Komaba,S.Adv.Energy Mater.2014, doi:10.1002/aenm.201301453 (26)Billaud,J.;Singh,G.;Armstrong,A.R.;Gonzalo,E.; Roddatis,V.;Armand,M.;Rojo,T.;Bruce,P.G.Energy Environ.Sci.2014,7,1387.doi:10.1039/c4ee00465e (27)Yabuuchi,N.;Hara,R.;Kubota,K.;Paulsen,J.;Kumakura,S.; Komaba,S.J.Mater.Chem.A 2014,2,16851.doi:10.1039/ c4ta04351k (28)Billaud,J.;Clément,R.J.;Armstrong,A.R.;Canales-Vázquez,J.;Rozier,P.;Grey,C.P.;Bruce,P.G.J.Am.Chem. Soc.2014,136,17243.doi:10.1021/ja509704t (29)Yabuuchi,N.;Yoshida,H.;Komaba,S.Electrochemistry 2012, 80,716.doi:10.5796/electrochemistry.80.716 (30)Zhao,J.;Zhao,L.;Dimov,N.;Okada,S.;Nishida,T.J. Electrochem.Soc.2013,160,A3077.doi:10.1149/2.007305jes (31)Lee,E.;Brown,D.E.;Alp,E.E.;Ren,Y.;Lu,J.;Woo,J.J.; Johnson,C.S.Chem.Mater.2015,27,6755.doi:10.1021/acs. chemmater.5b02918 (32)Yabuuchi,N.;Kajiyama,M.;Iwatate,J.;Nishikawa,H.; Hitomi,S.;Okuyama,R.;Usui,R.;Yamada,Y.;Komaba,S. Nat.Mater.2012,11,512.doi:10.1038/nmat3309 (33)Yuan,D.;Hu,X.;Qian,J.;Pei,F.;Wu,F.;Mao,R.;Ai,X.; Yang,H.;Cao,Y.Electrochim.Acta 2014,116,300. doi:10.1016/j.electacta.2013.10.211 (34)Thorne,J.S.;Dunlap,R.A.;Obrovac,M.N.J.Electrochem. Soc.2013,160,A361.doi:10.1149/2.058302jes (35)Singh,G.;Acebedo,B.;Cabanas,M.C.;Shanmukaraj,D.; Armand,M.;Rojo,T.Electrochem.Commun.2013,37,61. doi:10.1016/j.elecom.2013.10.008 (36)Wang,X.;Liu,G.;Iwao,T.;Okubo,M.;Yamada,A.J.Phys. Chem.C 2014,118,2970.doi:10.1021/jp411382r (37)Singh,G.;Aguesse,F.;Otaegui,L.;Goikolea,E.;Gonzalo,E.; Segalini,J.;Rojo,T.J.Power Sources 2015,273,333.doi:10.1016/j.jpowsour.2014.09.050 (38)(a)Miyazaki,S.;Kikkawa,S.;Koizumi,M.Synth.Met.1983, 6,211.doi:10.1016/0379-6779(83)90156-X (b)Molenda,J.;StokŁlosa,A.Solid State Ionics 1990,38,1. doi:10.1016/0167-2738(90)90438-W (39)Vassilaras,P.;Ma,X.;Li,X.;Ceder,G.J.Electrochem.Soc. 2013,160,A207.doi:10.1149/2.023302jes (40)Han,M.H.;Gonzalo,E.;Casas-Cabanas,M.;Rojo,T.J. Power Sources 2014,258,266.doi:10.1016/j. jpowsour.2014.02.048 (41)(a)Komaba,S.;Yabuuchi,N.;Nakayama,T.;Ogata,A.; Ishikawa,T.;Nakai,I.Inorg.Chem.2012,51,6211. doi:10.1021/ic300357d (b)Wang,H.;Yang,B.;Liao,X.Z.;Xu,J.;Yang,D.;He,Y.S.; Ma,Z.F.Electrochim.Acta 2013,113,200.doi:10.1016/j. electacta.2013.09.098 (42)Yuan,D.D.;Wang,Y.X.;Cao,Y.L.;Ai,X.P.;Yang,H.X. ACS Appl.Mater.Interfaces 2015,7,8585.doi:10.1021/ acsami.5b00594 (43)Yoshida,H.;Yabuuchi,N.;Kubota,K.;Ikeuchi,I.;Garsuch, A.;Schulz-Dobrick,M.;Komaba,S.Chem.Commun.2014, 50,3677.doi:10.1039/c3cc49856e (44)Yu,H.;Guo,S.;Zhu,Y.;Ishida,M.;Zhou,H.Chem.Commun. 2014,50,457.doi:10.1039/c3cc47351a (45)Shanmugam,R.;Lai,W.ECS Electrochem.Lett.2014,3,A23. doi:10.1149/2.007404eel (46)Gupta,A.;Buddie Mullins,C.;Goodenough,J.B.J.Power Sources 2013,243,817.doi:10.1016/j.jpowsour.2013.06.073 (47)Saadoune,I.;Maazaz,A.;Ménétrier,M.;Delmas,C.J.Solid State Chem.1996,122,111.doi:10.1006/jssc.1996.0090 (48)Kim,D.;Kang,S.H.;Slater,M.;Rood,S.;Vaughey,J.T.; Karan,N.;Balasubramanian,M.;Johnson,C.S.Adv.Energy Mater.2011,1,333.doi:10.1002/aenm.201000061 (49)(a)Guo,S.;Liu,P.;Yu,H.;Zhu,Y.;Chen,M.;Ishida,M.; Zhou,H.Angew.Chem.2015,127,5992.doi:10.1002/ ange.201411788 (b)Lee,E.;Lu,J.;Ren,Y.;Luo,X.;Zhang,X.;Wen,J.;Miller, D.;DeWahl,A.;Hackney,S.;Key,B.;Kim,D.;Slater,M.D.; Johnson,C.S.Adv.Energy Mater.2014,doi:10.1002/ aenm.201400458 (50)Yuan,D.;Liang,X.;Wu,L.;Cao,Y.;Ai,X.;Feng,J.;Yang,H. Adv.Mater.2014,26,6301.doi:10.1002/adma.201401946 (51)Ma,J.;Bo,S.H.;Wu,L.;Zhu,Y.;Grey,C.P.;Khalifah,P.G. Chem.Mater.2015,27,2387.doi:10.1021/cm504339y (52)(a)Mu,L.;Xu,S.;Li,Y.;Hu,Y.S.;Li,H.;Chen,L.;Huang, X.Adv.Mater.2015,27,6928.doi:10.1002/adma.201502449 (b)Xu,S.Y.;Wu,X.Y.;Li,Y.M.;Hu,Y.S.;Chen,L.Q.Chin. Phys.B 2014,23,118202.doi; (c)Li,Y.;Yang,Z.;Xu,S.;Mu,L.;Gu,L.;Hu,Y.S.;Li,H.; Chen,L.Adv.Sci.2015,doi:10.1002/advs.201500031 (d)Li,Y.;Hu,Y.S.;Qi,X.;Rong,X.;Li,H.;Huang,X.;Chen, L.Energy Storage Materials 2016,5,191.doi:10.1016/j. ensm.2016.07.006 (53)Berthelot,R.;Carlier,D.;Delmas,C.Nat.Mater.2011,10,74. doi:10.1038/nmat2920 (54)Ding,J.J.;Zhou,Y.N.;Sun,Q.;Yu,X.Q.;Yang,X.Q.;Fu,Z. W.Electrochim.Acta 2013,87,388.doi:10.1016/j. electacta.2012.09.058 (55)Ma,Y.;Doeff,M.M.;Visco,S.J.;De Jonghe,L.C.J. Electrochem.Soc.1993,140,2726.doi:10.1149/1.2220900 (56)Yoshida,H.;Yabuuchi,N.;Komaba,S.Electrochem.Commun. 2013,34,60.doi:10.1016/j.elecom.2013.05.012 (57)Carlier,D.;Cheng,J.H.;Berthelot,R.;Guignard,M.; Yoncheva,M.;Stoyanova,R.;Hwang,B.J.;Delmas,C. Dalton Trans.2011,40,9306.doi:10.1039/c1dt10798d (58)(a)Chagas,L.;Buchholz,D.;Vaalma,C.;Wu,L.;Passerini,S. J.Mater.Chem.A 2014,2,20263.doi:10.1039/C4TA03946G (b)Chen,X.;Zhou,X.;Hu,M.;Liang,J.;Wu,D.;Wei,J.; Zhou,Z.,J.Mater.Chem.A 2015,3,20708.doi:10.1039/ C5TA05205J (59)Komaba,S.;Takei,C.;Nakayama,T.;Ogata,A.;Yabuuchi,N. Electrochem.Commun.2010,12,355.doi:10.1016/j. elecom.2009.12.033 (60)Ding,J.J.;Zhou,Y.N.;Sun,Q.;Fu,Z.W.Electrochem. Commun.2012,22,85.doi:10.1016/j.elecom.2012.06.001 (61)(a)Nohira,T.;Ishibashi,T.;Hagiwara,R.J.Power Sources 2012,205,506.doi:10.1016/j.jpowsour.2011.11.086 (b)Chen,C.Y.;Matsumoto,K.;Nohira,T.;Hagiwara,R.; Fukunaga,A.;Sakai,S.;Nitta,K.;Inazawa,S.J.Power Sources 2013,237,52.doi:10.1016/j.jpowsour.2013.03.006 (62)(a)Zhou,Y.N.;Ding,J.J.;Nam,K.W.;Yu,X.;Bak,S.M.; Hu,E.;Liu,J.;Bai,J.;Li,H.;Fu,Z.W.;Yang,X.Q.J.Mater. Chem.A 2013,1,11130.doi:10.1039/c3ta12282d (b)Kubota,K.;Ikeuchi,I.;Nakayama,T.;Takei,C.;Yabuuchi, N.;Shiiba,H.;Nakayama,M.;Komaba,S.J.Phys.Chem.C 2015,119,166.doi:10.1021/jp5105888 (c)Bo,S.H.;Li,X.;Toumar,A.J.;Ceder,G.Chem.Mater. 2016,doi:10.1021/acs.chemmater.5b04626 (63)Yu,C.Y.;Park,J.S.;Jung,H.G.;Chung,K.Y.;Aurbach,D.; Sun,Y.K.;Myung,S.T.Energy Environ.Sci.2015,8,2019. doi:10.1039/c5ee00695c (64)Wang,Y.;Xiao,R.;Hu,Y.S.;Avdeev,M.;Chen,L.Nat. Commun.2015,6.doi:10.1038/ncomms7954 (65)(a)Masashige,O.J.Phys.:Condens.Matter 2008,20,145205. doi:10.1088/0953-8984/20/14/145205 (b)McQueen,T.M.;Stephens,P.W.;Huang,Q.;Klimczuk, T.;Ronning,F.;Cava,R.J.Phys.Rev.Lett.2008,101,166402. doi:10.1103/PhysRevLett.101.166402 (66)(a)Guignard,M.;Didier,C.;Darriet,J.;Bordet,P.;Elkaïm,E.; Delmas,C.Nat.Mater.2013,12,74.doi:10.1038/nmat3478 (b)Didier,C.;Guignard,M.;Darriet,J.;Delmas,C.Inorg. Chem.2012,51,11007.doi:10.1021/ic301505e (67)Hamani,D.;Ati,M.;Tarascon,J.M.;Rozier,P.Electrochem. Commun.2011,13,938.doi:10.1016/j.elecom.2011.06.005 (68)Masquelier,C.;Croguennec,L.Chem.Rev.2013,113,6552. doi:10.1021/cr3001862 (69)(a)Zaghib,K.;Trottier,J.;Hovington,P.;Brochu,F.;Guerfi, A.;Mauger,A.;Julien,C.M.J.Power Sources 2011,196, 9612.doi:10.1016/j.jpowsour.2011.06.061 (b)Sun,A.;Manivannan,A.ECS Trans.2011,35,3. doi:10.1149/1.3655683 (c)Oh,S.M.;Myung,S.T.;Hassoun,J.;Scrosati,B.;Sun,Y. K.Electrochem.Commun.2012,22,149.doi:10.1016/j. elecom.2012.06.014 (70)Le Poul,N.;Baudrin,E.;Morcrette,M.;Gwizdala,S.; Masquelier,C.;Tarascon,J.M.Solid State Ionics 2003,159, 149.doi:10.1016/S0167-2738(02)00921-9 (71)Zhu,Y.;Xu,Y.;Liu,Y.;Luo,C.;Wang,C.Nanoscale 2013,5, 780.doi:10.1039/c2nr32758a (72)(a)Moreau,P.;Guyomard,D.;Gaubicher,J.;Boucher,F. Chem.Mater.2010,22,4126.doi:10.1021/cm101377h (b)Galceran,M.;Saurel,D.;Acebedo,B.;Roddatis,V.V.; Martin,E.;Rojo,T.;Casas-Cabanas,M.Phys.Chem.Chem. Phys.2014,16,8837.doi:10.1039/c4cp01089b (c)Casas-Cabanas,M.;Roddatis,V.V.;Saurel,D.;Kubiak,P.; Carretero-Gonzalez,J.;Palomares,V.;Serras,P.;Rojo,T.J. Mater.Chem.2012,22,17421.doi:10.1039/c2jm33639a (73)Lu,J.;Chung,S.C.;Nishimura,S.I.;Yamada,A.Chem.Mater. 2013,25,4557.doi:10.1021/cm402617b (74)Boucher,F.;Gaubicher,J.;Cuisinier,M.;Guyomard,D.; Moreau,P.J.Am.Chem.Soc.2014,136,9144.doi:10.1021/ ja503622y (75)Fang,Y.;Liu,Q.;Xiao,L.;Ai,X.;Yang,H.;Cao,Y.ACS Appl. Mater.Interfaces 2015,7,17977.doi:10.1021/ acsami.5b04691 (76)Uebou,Y.;Kiyabu,T.;Okada,S.;Yamaki,J.I.Rep.Inst.Adv. Mater.Study Kyushu Univ.2002,16,1.doi:10.15017/7951 (77)Jian,Z.;Zhao,L.;Pan,H.;Hu,Y.S.;Li,H.;Chen,W.;Chen, L.Electrochem.Commun.2012,14,86.doi:10.1016/j. elecom.2011.11.009 (78)(a)Duan,W.;Zhu,Z.;Li,H.;Hu,Z.;Zhang,K.;Cheng,F.; Chen,J.J.Mater.Chem.A 2014,2,8668.doi:10.1039/ c4ta00106k (b)Jian,Z.;Han,W.;Lu,X.;Yang,H.;Hu,Y.S.;Zhou,J.; Zhou,Z.;Li,J.;Chen,W.;Chen,D.;Chen,L.Adv.Energy Mater.2013,3,156.doi:10.1002/aenm.201200558 (c)Li,S.;Dong,Y.;Xu,L.;Xu,X.;He,L.;Mai,L.Adv.Mater. 2014,26,3545.doi:10.1002/adma.201305522 (d)Rui,X.;Sun,W.;Wu,C.;Yu,Y.;Yan,Q.Adv.Mater.2015, 27,6670.doi:10.1002/adma.201502864 (e)Jiang,Y.;Yang,Z.;Li,W.;Zeng,L.;Pan,F.;Wang,M.; Wei,X.;Hu,G.;Gu,L.;Yu,Y.Adv.Energy Mater.2015, doi:10.1002/aenm.201402104 (f)Li,H.;Bai,Y.;Wu,F.;Li,Y.;Wu,C.J.Power Sources 2015,273,784.doi:10.1016/j.jpowsour.2014.09.153 (79)(a)Jian,Z.;Yuan,C.;Han,W.;Lu,X.;Gu,L.;Xi,X.;Hu,Y. S.;Li,H.;Chen,W.;Chen,D.;Ikuhara,Y.;Chen,L.Adv. Funct.Mater.2014,24,4265.doi:10.1002/adfm.201400173 (b)Lim,S.Y.;Kim,H.;Shakoor,R.A.;Jung,Y.;Choi,J.W.J. Electrochem.Soc.2012,159,A1393.doi:10.1149/2.015209jes (80)Saravanan,K.;Mason,C.W.;Rudola,A.;Wong,K.H.; Balaya,P.Adv.Energy Mater.2013,3,444.doi:10.1002/ aenm.201200803 (81)Zhu,C.;Song,K.;vanAken,P.A.;Maier,J.;Yu,Y.Nano Lett. 2014,14,2175.doi:10.1021/nl500548a (82)(a)Zhu,C.;Kopold,P.;vanAken,P.A.;Maier,J.;Yu,Y.Adv. Mater.2016,doi:10.1002/adma.201505943 (b)Plashnitsa,L.S.;Kobayashi,E.;Noguchi,Y.;Okada,S.; Yamaki,J.I.J.Electrochem.Soc.2010,157,A536. doi:10.1149/1.3298903 (83)Fang,Y.;Xiao,L.;Ai,X.;Cao,Y.;Yang,H.Adv.Mater.2015, 27,5895.doi:10.1002/adma.201502018 (84)Fang,Y.;Xiao,L.;Qian,J.;Cao,Y.;Ai,X.;Huang,Y.;Yang, H.Adv.Energy Mater.2016,doi:10.1002/aenm.201502197 (85)Mason,C.W.;Gocheva,I.;Hoster,H.E.;Yu,D.Y.W.ECS Trans.2014,58,41.doi:10.1149/05812.0041ecst (86)Lim,S.J.;Han,D.W.;Nam,D.H.;Hong,K.S.;Eom,J.Y.; Ryu,W.H.;Kwon,H.S.J.Mater.Chem.A 2014,2,19623. doi:10.1039/c4ta03948c (87)Li,H.;Yu,X.;Bai,Y.;Wu,F.;Wu,C.;Liu,L.Y.;Yang,X.Q. J.Mater.Chem.A 2015,3,9578.doi:10.1039/c5ta00277j (88)Barpanda,P.;Ye,T.;Nishimura,S.I.;Chung,S.C.;Yamada, Y.;Okubo,M.;Zhou,H.;Yamada,A.Electrochem.Commun. 2012,24,116.doi:10.1016/j.elecom.2012.08.028 (89)Barpanda,P.;Liu,G.;Ling,C.D.;Tamaru,M.;Avdeev,M.; Chung,S.C.;Yamada,Y.;Yamada,A.Chem.Mater.2013,25, 3480.doi:10.1021/cm401657c (90)Kim,H.;Shakoor,R.A.;Park,C.;Lim,S.Y.;Kim,J.S.;Jo,Y. N.;Cho,W.;Miyasaka,K.;Kahraman,R.;Jung,Y.;Choi,J. W.Adv.Funct.Mater.2013,23,1147.doi:10.1002/ adfm.201201589 (91)(a)Park,C.S.;Kim,H.;Shakoor,R.A.;Yang,E.;Lim,S.Y.; Kahraman,R.;Jung,Y.;Choi,J.W.J.Am.Chem.Soc.2013, 135,2787.doi:10.1021/ja312044k (b)Barpanda,P.;Ye,T.;Avdeev,M.;Chung,S.C.;Yamada, A.J.Mater.Chem.A 2013,1,4194.doi:10.1039/c3ta10210f (92)(a)Barpanda,P.;Liu,G.;Mohamed,Z.;Ling,C.D.;Yamada, A.Solid State Ion.2014,268,Part B,305.doi:10.1016/j. ssi.2014.03.011 (b)Tealdi,C.;Ricci,M.;Ferrara,C.;Bruni,G.;Quartarone,E.; Mustarelli,P.Batteries 2016,2,1.doi:10.3390/ batteries2010001 (c)Shakoor,R.A.;Park,C.S.;Raja,A.A.;Shin,J.; Kahraman,R.Phys.Chem.Chem.Phys.2016,18,3929.doi:10.1039/c5cp06836c (93)Barpanda,P.;Avdeev,M.;Ling,C.D.;Lu,J.;Yamada,A. Inorg.Chem.2013,52,395.doi:10.1021/ic302191d (94)Barpanda,P.;Liu,G.;Avdeev,M.;Yamada,A. ChemElectroChem 2014,1,1488.doi:10.1002/ celc.201402095 (95)Kim,J.;Park,I.;Kim,H.;Park,K.Y.;Park,Y.U.;Kang,K. Adv.Energy Mater.2016,doi:10.1002/aenm.201502147 (96)Barpanda,P.;Oyama,G.;Nishimura,S.I.;Chung,S.C.; Yamada,A.Nat.Commun.2014,5.doi:10.1038/ncomms5358 (97)Oyama,G.;Nishimura,S.I.;Suzuki,Y.;Okubo,M.;Yamada, A.ChemElectroChem 2015,2,1019.doi:10.1002/ celc.201500036 (98)Meng,Y.;Yu,T.;Zhang,S.;Deng,C.J.Mater.Chem.A 2016, 4,1624.doi:10.1039/c5ta07696j (99)Wei,S.;Mortemard de Boisse,B.;Oyama,G.;Nishimura,S. I.;Yamada,A.ChemElectroChem 2016,3,209.doi:10.1002/ celc.201500455 (100)Singh,P.;Shiva,K.;Celio,H.;Goodenough,J.B.Energy Environ.Sci.2015,8,3000.doi:10.1039/c5ee02274f (101)(a)Dwibedi,D.;Araujo,R.B.;Chakraborty,S.;Shanbogh,P. P.;Sundaram,N.G.;Ahuja,R.;Barpanda,P.J.Mater.Chem.A 2015,3,18564.doi:10.1039/c5ta04527d (b)Reynaud,M.;Rousse,G.;Abakumov,A.M.;Sougrati,M. T.;Van Tendeloo,G.;Chotard,J.N.;Tarascon,J.M.J.Mater. Chem.A 2014,2,2671.doi:10.1039/c3ta13648e (c)Araujo,R.B.;Islam,M.S.;Chakraborty,S.;Ahuja,R.J. Mater.Chem.A 2016,4,451.doi:10.1039/c5ta08114a (102)(a)Padhi,A.K.;Nanjundaswamy,K.S.;Masquelier,C.; Okada,S.;Goodenough,J.B.J.Electrochem.Soc.1997,144, 1609.doi:10.1149/1.1837649 (b)Padhi,A.K.;Manivannan,V.;Goodenough,J.B.J. Electrochem.Soc.1998,145,1518.doi:10.1149/1.1838513 (103)Ellis,B.L.;Makahnouk,W.R.M.;Makimura,Y.;Toghill,K.; Nazar,L.F.Nat.Mater.2007,6,749.doi:10.1038/nmat2007 (104)Recham,N.;Chotard,J.N.;Dupont,L.;Djellab,K.;Armand, M.;Tarascon,J.M.J.Electrochem.Soc.2009,156,A993. doi:10.1149/1.3236480 (105)(a)Kawabe,Y.;Yabuuchi,N.;Kajiyama,M.;Fukuhara,N.; Inamasu,T.;Okuyama,R.;Nakai,I.;Komaba,S.Electrochem. Commun.2011,13,1225.doi:10.1016/j.elecom.2011.08.038 (b)Langrock,A.;Xu,Y.;Liu,Y.;Ehrman,S.;Manivannan,A.; Wang,C.J.Power Sources 2013,223,62.doi:10.1016/j. jpowsour.2012.09.059 (106)Le Meins,J.M.;Crosnier-Lopez,M.P.;Hemon-Ribaud,A.; Courbion,G.J.Solid State Chem.1999,148,260.doi:10.1006/ jssc.1999.8447 (107)(a)Gover,R.K.B.;Bryan,A.;Burns,P.;Barker,J.Solid State Ion.2006,177,1495.doi:10.1016/j.ssi.2006.07.028 (b)Barker,J.;Gover,R.K.B.;Burns,P.;Bryan,A.J. Electrochem.Solid-State Lett.2006,9,A190.doi:10.1149/ 1.2168288 (c)Song,W.;Liu,S.Solid State Sci.2013,15,1.doi:10.1016/ j.solidstatesciences.2012.09.012 (108)Shakoor,R.A.;Seo,D.H.;Kim,H.;Park,Y.U.;Kim,J.;Kim, S.W.;Gwon,H.;Lee,S.;Kang,K.J.Mater.Chem.2012,22, 20535.doi:10.1039/c2jm33862a (109)(a)Liu,Z.;Hu,Y.Y.;Dunstan,M.T.;Huo,H.;Hao,X.;Zou, H.;Zhong,G.;Yang,Y.;Grey,C.P.Chem.Mater.2014,26, 2513.doi:10.1021/cm403728w (b)Bianchini,M.;Fauth,F.;Brisset,N.;Weill,F.;Suard,E.; Masquelier,C.;Croguennec,L.Chem.Mater.2015,27,3009. doi:10.1021/acs.chemmater.5b00361 (110)Massa,W.;Yakubovich,O.V.;Dimitrova,O.V.Solid State Sci. 2002,4,495.doi:10.1016/S1293-2558(02)01283-9 (111)Sauvage,F.;Quarez,E.;Tarascon,J.M.;Baudrin,E.Solid State Sci.2006,8,1215.doi:10.1016/j. solidstatesciences.2006.05.009 (112)Serras,P.;Palomares,V.;Goni,A.;Gil de Muro,I.;Kubiak,P.; Lezama,L.;Rojo,T.J.Mater.Chem.2012,22,22301. doi:10.1039/c2jm35293a (113)Peng,M.;Li,B.;Yan,H.;Zhang,D.;Wang,X.;Xia,D.;Guo, G.Angew.Chem.Int.Ed.2015,54,6452.doi:10.1002/ anie.201411917 (114)Qi,Y.;Mu,L.;Zhao,J.;Hu,Y.S.;Liu,H.;Dai,S.Angew. Chem.2015,127,10049.doi:10.1002/ange.201503188 (115)(a)Park,Y.U.;Seo,D.H.;Kim,H.;Kim,J.;Lee,S.;Kim,B.; Kang,K.Adv.Funct.Mater.2014,24,4603.doi:10.1002/ adfm.201400561 (b)Xu,M.;Xiao,P.;Stauffer,S.;Song,J.;Henkelman,G.; Goodenough,J.B.Chem.Mater.2014,26,3089.doi:10.1021/ cm500106w (c)Park,Y.U.;Seo,D.H.;Kwon,H.S.;Kim,B.;Kim,J.; Kim,H.;Kim,I.;Yoo,H.I.;Kang,K.J.Am.Chem.Soc.2013, 135,13870.doi:10.1021/ja406016j (d)Serras,P.;Palomares,V.;Alonso,J.;Sharma,N.;López del Amo,J.M.;Kubiak,P.;Fdez-Gubieda,M.L.;Rojo,T.Chem. Mater.2013,25,4917.doi:10.1021/cm403679b (e)Sharma,N.;Serras,P.;Palomares,V.;Brand,H.E.A.; Alonso,J.;Kubiak,P.;Fdez-Gubieda,M.L.;Rojo,T.Chem. Mater.2014,26,3391.doi:10.1021/cm5005104 (116)(a)Barpanda,P.;Chotard,J.N.;Recham,N.;Delacourt,C.; Ati,M.;Dupont,L.;Armand,M.;Tarascon,J.M.Inorg.Chem. 2010,49,7401.doi:10.1021/ic100583f (b)Tripathi,R.;Gardiner,G.R.;Islam,M.S.;Nazar,L.F. Chem.Mater.2011,23,2278.doi:10.1021/cm200683n (c)Ellis,B.L.;Makahnouk,W.R.M.;Rowan-Weetaluktuk, W.N.;Ryan,D.H.;Nazar,L.F.Chem.Mater.2010,22,1059. doi:10.1021/cm902023h (117)Kim,H.;Park,I.;Seo,D.H.;Lee,S.;Kim,S.W.;Kwon,W.J.; Park,Y.U.;Kim,C.S.;Jeon,S.;Kang,K.J.Am.Chem.Soc. 2012,134,10369.doi:10.1021/ja3038646 (118)Kim,H.;Park,I.;Lee,S.;Kim,H.;Park,K.Y.;Park,Y.U.; Kim,H.;Kim,J.;Lim,H.D.;Yoon,W.S.;Kang,K.Chem. Mater.2013,25,3614.doi:10.1021/cm4013816 (119)Nose,M.;Nakayama,H.;Nobuhara,K.;Yamaguchi,H.; Nakanishi,S.;Iba,H.J.Power Sources 2013,234,175. doi:10.1016/j.jpowsour.2013.01.162 (120)Nose,M.;Shiotani,S.;Nakayama,H.;Nobuhara,K.; Nakanishi,S.;Iba,H.Electrochem.Commun.2013,34,266. doi:10.1016/j.elecom.2013.07.004 (121)Wood,S.M.;Eames,C.;Kendrick,E.;Islam,M.S.J.Phys. Chem.C 2015,119,15935.doi:10.1021/acs.jpcc.5b04648 (122)(a)Deng,C.;Zhang,S.ACS Appl.Mater.Interfaces 2014,6, 9111.doi:10.1021/am501072j (b)Zhang,S.;Deng,C.;Meng,Y.J.Mater.Chem.A 2014,2, 20538.doi:10.1039/c4ta04499a (123)(a)Hautier,G.;Jain,A.;Chen,H.;Moore,C.;Ong,S.P.; Ceder,G.J.Mater.Chem.2011,21,17147.doi:10.1039/ c1jm12216a (b)Jain,A.;Hautier,G.;Moore,C.;Kang,B.;Lee,J.;Chen, H.;Twu,N.;Ceder,G.J.Electrochem.Soc.2012,159,A622. doi:10.1149/2.080205jes (124)Chen,H.;Hao,Q.;Zivkovic,O.;Hautier,G.;Du,L.S.;Tang, Y.;Hu,Y.Y.;Ma,X.;Grey,C.P.;Ceder,G.Chem.Mater. 2013,25,2777.doi:10.1021/cm400805q (125)Huang,W.;Zhou,J.;Li,B.;Ma,J.;Tao,S.;Xia,D.;Chu,W.; Wu,Z.Sci.Rep.2014,4,4188.doi:10.1038/srep04188 (126)Qian,J.;Zhou,M.;Cao,Y.;Ai,X.;Yang,H.Adv.Energy Mater.2012,2,410.doi:10.1002/aenm.201100655 (127)Qian,J.;Zhou,M.;Cao,Y.J.Electrochem 2012,18,108. (128)Lu,Y.;Wang,L.;Cheng,J.;Goodenough,J.B.Chem. Commun.2012,48,6544.doi:10.1039/C2CC31777J (129)Wu,X.;Deng,W.;Qian,J.;Cao,Y.;Ai,X.;Yang,H.J.Mater. Chem.A 2013,1,10130.doi:10.1039/c3ta12036h (130)Liu,Y.;Qiao,Y.;Zhang,W.;Li,Z.;Ji,X.;Miao,L.;Yuan,L.; Hu,X.;Huang,Y.Nano Energy 2015,12,386.doi:10.1016/j. nanoen.2015.01.012 (131)You,Y.;Wu,X.L.;Yin,Y.X.;Guo,Y.G.Energy Environ.Sci. 2014,7,1643.doi:10.1039/c3ee44004d (132)You,Y.;Yu,X.;Yin,Y.;Nam,K.W.;Guo,Y.G.Nano Res. 2014,8,117.doi:10.1007/s12274-014-0588-7 (133)(a)Wessells,C.D.;Peddada,S.V.;Huggins,R.A.;Cui,Y. Nano Lett.2011,11,5421.doi:10.1021/nl203193q (b)Wang,L.;Song,J.;Qiao,R.;Wray,L.A.;Hossain,M.A.; Chuang,Y.D.;Yang,W.;Lu,Y.;Evans,D.;Lee,J.J.;Vail,S.; Zhao,X.;Nishijima,M.;Kakimoto,S.;Goodenough,J.B.J. Am.Chem.Soc.2015,137,2548.doi:10.1021/ja510347s (134)Wang,L.;Lu,Y.;Liu,J.;Xu,M.;Cheng,J.;Zhang,D.; Goodenough,J.B.Angew.Chem.Int.Ed.2013,52,1964. doi:10.1002/anie.201206854 (135)Song,J.;Wang,L.;Lu,Y.;Liu,J.;Guo,B.;Xiao,P.;Lee,J.J.; Yang,X.Q.;Henkelman,G.;Goodenough,J.B.J.Am.Chem. Soc.2015,137,2658.doi:10.1021/ja512383b (136)Yang,D.;Xu,J.;Liao,X.Z.;He,Y.S.;Liu,H.;Ma,Z.F. Chem.Commun.2014,50,13377.doi:10.1039/c4cc05830e (137)Li,W.J.;Chou,S.L.;Wang,J.Z.;Wang,J.L.;Gu,Q.F.;Liu, H.K.;Dou,S.X.Nano Energy 2015,13,200.doi:10.1016/j. nanoen.2015.02.019 (138)Lee,E.;Kim,D.H.;Hwang,J.;Kang,J.S.;Van Minh,N.; Yang,I.S.;Ueno,T.;Sawada,M.J.Korean Phys.Soc.2013, 62,1910.doi:10.3938/jkps.62.1910 (139)Masamitsu,T.;Tomoyuki,M.;Yutaka,M.Appl.Phys.Express 2013,6,025802.doi:10.7567/APEX.6.025802 (140)Xie,M.;Xu,M.;Huang,Y.;Chen,R.;Zhang,X.;Li,L.;Wu, F.Electrochem.Commun.2015,59,91.doi:10.1016/j. elecom.2015.07.014 (141)Wu,X.;Wu,C.;Wei,C.;Hu,L.;Qian,J.;Cao,Y.;Ai,X.; Wang,J.;Yang,H.ACS Appl.Mater.Interfaces 2016,8,5393. doi:10.1021/acsami.5b12620 (142)You,Y.;Wu,X.L.;Yin,Y.X.;Guo,Y.G.J.Mater.Chem.A 2013,1,14061.doi:10.1039/c3ta13223d (143)Yue,Y.;Binder,A.J.;Guo,B.;Zhang,Z.;Qiao,Z.A.;Tian, C.;Dai,S.Angew.Chem.2014,126,3198.doi:10.1002/ ange.201310679 (144)Okubo,M.;Li,C.H.;Talham,D.R.Chem.Commun.2014, 50,1353.doi:10.1039/c3cc47607c (145)Guo,C.;Zhang,K.;Zhao,Q.;Pei,L.;Chen,J.Chem. Commun.2015,51,10244.doi:10.1039/c5cc02251g (146)Luo,W.;Allen,M.;Raju,V.;Ji,X.Adv.Energy Mater.2014, doi:10.1002/aenm.201400554 (147)Deng,W.;Shen,Y.;Qian,J.;Cao,Y.;Yang,H.ACS Appl. Mater.Interfaces 2015,7,21095.doi:10.1021/ acsami.5b04325 (148)Yao,M.;Kuratani,K.;Kojima,T.;Takeichi,N.;Senoh,H.; Kiyobayashi,T.Sci.Rep.2014,4,3650.doi:10.1038/ srep03650 (149)Chihara,K.;Chujo,N.;Kitajou,A.;Okada,S.Electrochim. Acta 2013,110,240.doi:10.1016/j.electacta.2013.04.100 (150)Wang,S.;Wang,L.;Zhu,Z.;Hu,Z.;Zhao,Q.;Chen,J.Angew. Chem.In.Ed.2014,53,5892.doi:10.1002/anie.201400032 (151)Zhao,R.;Zhu,L.;Cao,Y.;Ai,X.;Yang,H.X.Electrochem. Commun.2012,21,36.doi:10.1016/j.elecom.2012.05.015 (152)Zhou,M.;Zhu,L.;Cao,Y.;Zhao,R.;Qian,J.;Ai,X.;Yang,H. RSC Adv.2012,2,5495.doi:10.1039/c2ra20666h (153)Zhou,M.;Xiong,Y.;Cao,Y.;Ai,X.;Yang,H.J.Polym.Sci., Part B:Polym.Phys.2013,51,114.doi:10.1002/polb.23184 (154)Zhu,L.;Shen,Y.;Sun,M.;Qian,J.;Cao,Y.;Ai,X.;Yang,H. Chem.Commun.2013,49,11370.doi:10.1039/c3cc46642f (155)Wang,H.G.;Yuan,S.;Ma,D.L.;Huang,X.L.;Meng,F.L.; Zhang,X.B.Adv.Energy Mater.2014,doi:10.1002/ aenm.201301651 (156)Xu,F.;Xia,J.;Shi,W.Electrochem.Commun.2015,60,117. doi:10.1016/j.elecom.2015.08.027 (157)Xu,F.;Xia,J.;Shi,W.;Cao,S.A.Mater.Chem.Phys.2016, 169,192.doi:10.1016/j.matchemphys.2015.12.004 (158)Dai,Y.;Zhang,Y.;Gao,L.;Xu,G.;Xie,J.Electrochem.Solid-State Lett.2010,13,A22.doi:10.1149/1.3276736 (159)Deng,W.;Liang,X.;Wu,X.;Qian,J.;Cao,Y.;Ai,X.;Feng, J.;Yang,H.Sci.Rep.2013,3,2671.doi:10.1038/srep02671 (160)Banda,H.;Damien,D.;Nagarajan,K.;Hariharan,M.; Shaijumon,M.M.J.Mater.Chem.A 2015,3,10453. doi:10.1039/C5TA02043C (161)Shiratsuchi,T.;Okada,S.;Yamaki,J.;Nishida,T.J.Power Sources 2006,159,268.doi:10.1016/j.jpowsour.2006.04.047 (162)Zhao,J.;Jian,Z.;Ma,J.;Wang,F.;Hu,Y.S.;Chen,W.;Chen, L.;Liu,H.;Dai,S.ChemSusChem 2012,5,1495.doi:10.1002/ cssc.201100844 (163)Liu,Y.;Xu,Y.;Han,X.;Pellegrinelli,C.;Zhu,Y.;Zhu,H.; Wan,J.;Chung,A.C.;Vaaland,O.;Wang,C.Nano Lett.2012, 12,5664.doi:10.1021/nl302819f (164)Fang,Y.;Xiao,L.;Qian,J.;Ai,X.;Yang,H.;Cao,Y.Nano Lett.2014,14,3539.doi:10.1021/nl501152f (165)Xu,S.;Zhang,S.;Zhang,J.;Tan,T.;Liu,Y.J.Mater.Chem.A 2014,2,7221.doi:10.1039/C4TA00239C (166)Wang,W.;Wang,S.;Jiao,H.;Zhan,P.;Jiao,S.Phys.Chem. Chem.Phys.2015,17,4551.doi:10.1039/C4CP05764C (167)Li,C.;Miao,X.;Chu,W.;Wu,P.;Tong,D.G.J.Mater.Chem. A 2015,3,8265.doi:10.1039/C5TA01191D (168)Uchaker,E.;Zheng,Y.;Li,S.;Candelaria,S.;Hu,S.;Cao,G. J.Mater.Chem.A 2014,2,18208.doi:10.1039/C4TA03788J (169)Fu,S.Y.;Li,Y.Z.;Chu,W.;Yang,Y.M.;Tong,D.G.;Le Zeng,Q.J.Mater.Chem.A 2015,3,16716.doi:10.1039/ C5TA04288G Recent Developments in Cathode Materials for Na Ion Batteries FANG Yong-Jin1CHEN Zhong-Xue2AI Xin-Ping1YANG Han-Xi1CAO Yu-Liang1,* (1College of Chemistry and Molecular Sciences,Wuhan University,Wuhan 430072,P.R.China;2School of Power and Mechanical Engineering,Wuhan University,Wuhan 430072,P.R.China) Sodium ion batteries(SIBs)have attracted increasing attention for energy storage systems because of abundant and low cost sodium resources.However,the large ionic radius of sodium and its slow electrochemical kinetics are the main obstacles for the development of suitable electrodes for high-performance SIBs.The development of high-performance cathode materials is the key to improving the energy density of SIBs and facilitating their commercialization.Herein,we review the latest advances and progress of cathode materials for SIBs,including transition metal oxides,polyanions,ferrocyanides,organic materials and polymers, and amorphous materials.Additionally,we have summarized our previous works in this area,explore the relationship between structure and electrochemical performance,and discuss effective ways to improve the reversibility,working potential and structural stability of these cathode materials. Sodium ion battery;Cathode material;Development;Sodium storage reaction; Electrochemical reaction mechanism O647 iew] 10.3866/PKU.WHXB201610111www.whxb.pku.edu.cn Received:August 9,2016;Revised:October 10,2016;Published online:October 11,2016. *Corresponding author.Email:ylcao@whu.edu.cn;Tel:+86-27-68754526. The project was supported by the National Key Research Program of China(2016YFB0901501)and National Natural Science Foundation of China (21373155,21333007,21273090). 国家重点研发计划(2016YFB0901501)和国家自然科学基金(21373155,21333007,21273090)资助项目 ©Editorial office ofActa Physico-Chimica Sinica








3 总结与展望
