固溶体光催化材料的研究进展
2017-03-10荆涛戴瑛
荆 涛 戴 瑛
(山东大学物理学院,山东大学晶体材料国家重点实验室,济南 250100)
固溶体光催化材料的研究进展
荆 涛 戴 瑛*
(山东大学物理学院,山东大学晶体材料国家重点实验室,济南 250100)
掺杂能够实现传统宽带隙半导体光催化材料的可见光响应,但引入的局域杂质能级易成为载流子的复合中心,降低材料的光催化活性。固溶体方法可以实现带隙和带边位置的精确调控,使材料的光吸收和氧化还原电位达到最佳平衡,是改善其光催化性能的有效方法。本文结合我们课题组近些年来的研究,从固溶体方法对半导体光催化材料带隙和带边位置的调控以及对载流子分离和迁移等性质的影响出发,概述了近年来该领域的最新研究进展,总结了固溶体方法在发展中所面临的主要问题,并对其发展趋势进行展望。
光催化材料;掺杂;固溶体;电子结构;载流子分离
1 引言
由于在解决能源短缺和环境污染问题方面的广泛应用前景,光催化技术引起了人们极大的研究兴趣1-13。通常认为,半导体光催化可以概括为三个主要过程:(1)半导体价带上的电子吸收光跃迁至导带,产生光生电子和空穴对;(2)光生电子和空穴从半导体内部向表面迁移;(3)光生电子和空穴在表面与吸附物分别发生氧化还原反应。一方面,由于宽的本征带隙,传统半导体光催化材料如 TiO2和 SrTiO3等的光响应位于紫外光区,因此这些材料只能吸收和利用很小一部分的太阳能。另一方面,在光催化反应过程中,很大比例的光生电子和空穴在半导体的内部或表面发生复合,以发光和发热的形式散发部分能量。因此,大部分光催化材料太阳能转换效率非常低,不能满足大规模应用的要求。为了改善这些问题,人们通常采用两种策略,一是采用金属和非金属掺杂14-16,引入本征缺陷17,染料敏化18和利用表面等离体子效应19-21等方法拓展材料的光响应范围。二是采用微结构调控22-24,形成异质结25,26和沉积助催化剂27,28等方法提高材料的量子效率。
掺杂是拓展材料光响应范围的有效手段,例如 , 采 用 过 渡 金 属 元 素 掺 杂 替 代 TiO2中 的 Ti4+离子,可以在其带隙中引入杂质能级,使其具有可见光区的催化活性29,30。然而,掺杂虽然能够提高可见光的吸收,但整体的光催化活性却难于得到明显的改善。这是由于掺杂引入的局域杂质能级易成为载流子的复合中心,降低材料的量子效率和光催化活性。并且,掺杂在减小带隙的同时也会降低光生载流子的氧化还原能力。为了实现带隙和带边位置的精确调控,人们也采用固溶体方法以提高半导体光催化材料的光催化活性31-35。该方法能够对材料的光响应范围和氧化还原电位进行连续调控,使有效光吸收和载流子氧化还原能力达到平衡,从而最大程度提高材料的光催化性能2,36。近几年,固溶 体 方 法 在光催化材料改性方面的应用非常普遍,为提高材料的光催化活性提供了一条重要途径。
本文将从固溶体方法对材料带隙和带边位置的调控以及对光生载流子分离和迁移性质的影响出发,概述固溶体光催化材料近年来的一些研究进展。对存在的一些问题进行总结,并对其发展趋势进行展望。
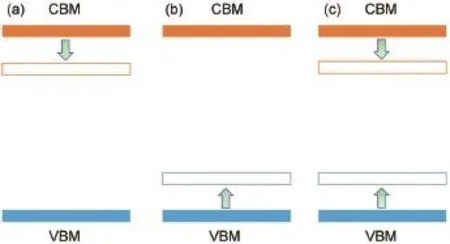
图1 固溶体方法调控带隙的三种策略Fig.1 Three strategies to tune band gap by solid solution method
2 固溶体方法调控材料的带隙和带边位置
固溶体方法可以实现带隙和带边位置的精确调控,对于实现光催化材料的改性具有重要意义。为了对半导体材料的带隙进行调控,如图1所示,可以从三个方面考虑:(1) 调控导带位置(a); (2) 调控价带位置(b);(3)同时调控价带和导带位置(c)。从电子结构来看,金属氧化物半导体材料的导带一般由金属元素未占据的d轨道或s轨道贡献,而价带一般由氧的p轨道或金属元素满占据的d轨道贡献。因此,采用两种导带位置不同的半导体构建固溶体,可以调控导带位置,而采用两种价带位置不同的半导体构建固溶体,可以调控价带位置,还可以采用价带和导带位置都不同的两种半导体构建固溶体,能够实现价带和导带位置的同时调控。

荆涛,1981年生。2010 年毕业于贵州大学理学院,获得硕士学位。2012 年至今就读于山东大学物理学院,攻读博士学位。主要从事光催化材料电子结构及其相关性质的研究。

戴瑛 ,1962 年 生。1998 年 博 士毕业于山东大学。现任山东大学物理学院教授,博士生导师。2015年被评为泰山学者特聘专家。主要从事半导体材料纳米光电性质和光催化性质及其应用的研究。
2.1 调控材料的导带位置
有些过渡金属氧化物半导体的导带主要由过渡金属未占据的d轨道贡献,引入同一族的过渡金属能够实现导带位置的调控。例如,AgTaO3是一种宽带隙光催化材料,带隙约为 3.4 eV,在牺牲剂的条件下,能够分别进行产氢和产氧半反应37。另一种半导体 AgNbO3的带隙约为 2.8 eV,由于 Nb 的4d轨道比Ta的5d轨道能量更低,该半导体的导带位置比 AgTaO3低。因此,AgNbO3不具备光催化分解水产氢的能力,但其产氧的活性较好。Ni等38采用水热法制 备了 AgTa1-xNbxO3固溶体,随着 Nb 浓度的增加,AgTa1-xNbxO3固溶体的带隙连续减小。在负载 NiO 助催化剂的情况下,AgTa1-xNbxO3(x= 0.3)能够在可见光照射下实现纯水的完全分解。Nb的混入降低了固溶体的导带位置,但其价带位置保 持 不 变39。 Zhou 等40采 用 水 热 结 晶 法 制 备 了Bi2MoxW1-xO6固溶体纳米片,发现随着 Mo 浓度的减小,纳米片的厚度也减小。可见光(> 400 nm)降解甲基蓝(MB)的实验表明,Bi2Mo0.25W0.75O6具有最高的光催化活性。而且,通过增加Mo的浓度能够调控该体系的带隙并提高可见光催化活性。Bi2WO6的 带 隙 约为 2.94 eV,能够 吸 收 很小部分的可见光。其价带主要由O的2p轨道和Bi的6s轨道贡献,而导带主要由W的5d轨道贡献。由于Mo的4d轨道能量低于W 的 5d轨道,因此通过增加Bi2MoxW1-xO6固溶体中 Mo 的浓度,可以降低导带位置实现材料带隙的减小,其 带 隙 能 够 在 2.94 到2.72 eV 范围内连续调控。
有些金属氧化物半导体的导带主要由金属元素的s和p轨道贡献,引入同族的元素也能够实现导带位置的调控。例如,Ye课题组41采用离子交换法处理 NaAl1-xGaxO2和 AgNO3,合成β-AgAl1-xGaxO2固溶体光催化材料,通过改变Ga的组分,其带隙能够在 2.19 到 2.83 eV 范围内连续调控,在 425 nm 左右的光照下,光降解异丙醇(iso-propanol)的量子效率可达到37.3%。电子结构的研究表明,固溶体的价带主要由Ag的4d和O的2p态贡献,而导带主要由Ag 的 5s5p态、Al的 3s3p态和 Ga的 4s4p态贡献。因此,固溶体随着Ga浓度的变化,价带位置保持不变,而导带位置连续下移(如图 2 所示)。这是由于 Ga的 4s4p轨道比 Al的 3s3p轨道能量更低,Ga的引入能够减小固溶体的带隙。Li等42采用溶胶-凝胶法合成的β-AgAl1-xGaxO2固溶体具有很好的结晶性,带隙能够在 2.31 到 2.7 eV 范围内调控。降解甲基橙的 实验表明,当x=0.8 时固溶体的光催化性 能 最 好 , 降 解 甲 基 橙 的 反 应 速 率 达 到 0.0402 min-1。 Ma 等43基 于 密 度 泛 函 理 论 研 究 了AgSb1-xBixO3固溶体光催化分解水的性质。发现通过增加 Bi的浓度,能够使固溶体的带隙从 2.6 eV减小到 2.0 eV。而且,在浓度x=0.21 时,固溶体将发生由焦绿石相(pyrochlore)到钛铁矿相(ilmenite)的转变。对于这两种不同的相,AgSb1-xBixO3固溶体的带隙都能够随着Bi组分的增加而减小。而当x=0.1875 时,由于氧化电位增强和带隙合适,焦绿石相的 AgSb1-xBixO3固溶体可能具有最好的光催化活性。AgSbO3的价带主要由 Ag 的 4d和 O 的2p轨道贡献,导带主要由Ag的5s态、O的2p态和Sb的 5s态贡献。而 AgSb1-xBixO3固溶体结构的导带主要 由 Ag 的 5s、O 的 2p、Sb 的 5s和 Bi的 6s态 构成,由于Bi―O键比Sb―O键更弱的相互作用,Bi的6s和O的2p反键轨道相互作用的程度也更弱,导致其导带位置比AgSbO3更低,因此随着Bi浓度的 增 加 , AgSb1-xBixO3固 溶 体 的 价 带 位 置 连 续 下移,带隙也逐渐减小。因此,利用同一族元素轨道能量的差异来改变固溶体光催化剂的导带位置,是实现带隙减小和可见光响应的有效手段。

图2 AgAlO2、AgGaO2和 AgAl1-xGaxO2固溶体的电子结构示意图Fig.2 Schematic electronic structures ofAgAlO2, AgGaO2andAgAl1-xGaxO2solid solutions
不同族元素的替换也能够实现固溶体材料导带位置的调控。例如 Yuan 等44采用改进的 Pechini-型工艺合成了 BaZr1-xSnxO3固溶体,在紫外光照射下能够分解水产生氢气和氧气。当Sn的组分x= 0.3 时产氢效率最高,析氢和析氧的反应速率分别为 138 和 37 µmol·h-1。 进 一 步 的 理 论 研 究 指 出 ,BaZrO3的导带主要由 Zr 4d态贡献,价带主要由 O 2p态贡献。随着 Sn 浓度的增加,Sn 5s态对固溶体导带的贡献逐渐增大,其带隙逐渐减小。同时,Sn 的引入使 BaZr1-xSnxO3固溶体发生了间接 带隙到直接带隙的转变,光吸收效率也得到有效地提高。
2.2 调控价带位置
一般来说,通过在金属氧化物半导体中引入p轨道能量更高的阴离子可以调控其价带位置。Umezawa 等45基 于 密 度 泛 函 理 论 研 究 了 TiO2(1-x)S2x固 溶 体, 由 于 S 3p轨 道 能量 明 显高 于 O 2p轨 道 ,价带位置的上移使固溶体的带隙明显减小,甚至在x低 于 0.25 时 , 固 溶 体 就 能 够 吸 收 可 见 光 。Li等46通过水热法,合成了 In(OH)ySz固溶体,能够在可见光驱动下降解丙酮气体。密度泛函理论的研究表明,In(OH)3为宽带隙半导体,带隙约为4.4 eV,其价带主要由 O 2p 轨道贡献。S 的混入使In(OH)ySz固溶体的价带位置上移,其导带位置几乎保持不变。进一步的研究表明,Zn掺杂的In(OH)ySz固溶体能够矿化丙酮和 RhB,使其可见光催化活性得到提高47,而氟化能够提高 In(OH)ySz固溶体降解 RhB 的光催化活性48。但由于金属氧化物半导体中阴离子替位的形成能较高,导致这些元素在固溶体中的溶解度较低,难以实现带隙和带边位置在较大范围内的连续调控。
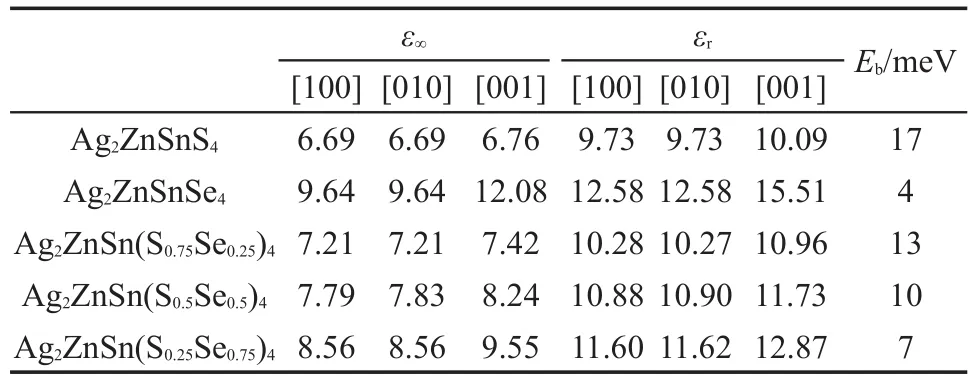
Domen 课题组通过氮化 Ga2O3和 ZnO 粉末合成了(GaN)x(ZnO)1-x固溶体49,能够在可见光照射下完全分解水。进一步的研究发现,以 Rh/Cr2O3为助催化剂,在 420-440 nm 范围光照下分解水的量子效率可以达到 5.9%50。通常,由两种不同半导体构建的固溶体结构,其带隙能够在二者的带隙值之间连续调控。而对于这一体系,构成固溶体的两种材料都为宽带隙半导体,只能吸收紫外光,其中GaN 的 带 隙 为 3.4 eV, 而 ZnO 为 3.2 eV。 有 趣 的是,当组分 x=0.13 时,(GaN)x(ZnO)1-x固溶体的带隙 减 小 到 2.58 eV。 密 度 泛 函 理 论 计 算 的 结 果 显示,固溶体的导带底主要由 Ga的 4s和4p轨道贡献,而价带顶主要由N的2p和Zn的3d轨道贡献。Zn的3d轨道与N的2p轨道之间强的排斥作用是导致固溶体价带位置上移和带隙减小的主要原因。用传统方法制备的(GaN)x(ZnO)1-x固溶体的 Zn 元素的溶解度较低(x<0.5),Huang 课题组51通过氮化层状双金属氢氧化物(包含 Zn2+和 Ga3+离子),制备了富 Zn 的(GaN)x(ZnO)1-x固溶体(0.46 图3 (ZnO)x(GaN)1-x固溶体光吸收谱(a)和带隙(b)随着Zn浓度的变化Fig.3 Light absorption spectra(a)and band gap(b)of (ZnO)x(GaN)1-xsolid solution with the change of Zn concentration 通过引入具有满占据d轨道的金属元素构建固溶体,可对光催化材料的带隙和带边位置具有非常明显的调控作用。例如,Maggard 课题组54用满足化学计量比的 LiNbO3、Cu2O 和 Nb2O5,通过固态反应法制备的 Li1-xCuxNb3O8固溶体光催化剂,能够在可见光照射下分解水产生氢气,随着Cu浓度的增加,固溶体的带隙能够在 3.89 到 1.27 eV 的范围内调控。由于 LiNb3O8的价带主要由 O 的 2p 轨道贡献,而导带主要由Nb的4d轨道贡献,碱金属Li对材料的能带边没有明显的贡献。由于Cu的3d轨道比 O 的 2p 轨道能量明显更高,Cu1+离子满占据3d 态的引入,使固溶体 Li1-xCuxNb3O8的价带位置抬高 , 材 料 的 带 隙 被 显 著 减 小 。Ye 课 题 组55采 用Ag2O、NaCO3和 Nb2O5等,通过固态反应法制备了Ag1-xNaxNbO3固 溶 体 光 催 化 剂 , 发 现 随 着 Na 组 分的增加,固溶体的带隙不断增大,光吸收范围也变窄。但价带位置下降使材料的氧化能力增强,当组分 x=0.4 时,该固溶体 的 光 催化活性最高。AgNbO3的价带主要由Ag的4d轨道和O的2p轨道贡献,而Na元素的引入减弱了Ag的4d轨道和O的 2p 轨道之间的排斥作用,使 Ag1-xNaxNbO3固溶体的价带位置下移,从而增强了其光生空穴的氧化能力。 Bi 6s态和O 2p态的耦合使Bi-基氧化物半导体具有较高的价带位置,因此Bi的引入能够提高固溶体的价 带 位 置 。 例 如 ,Zou 课 题 组56以 In2O3、Bi2O3和Ta2O5等为原料,采用固态反应法制备的 BixIn1-xTaO4固溶体,能够在可见光照射下分别进行产氢和产氧半反应,Bi组分 x=0.5 时的光催化活性明显高于 x=0.2 和 x=0.8。 第 一 性 原 理 计 算 结 果 表 明 ,InTaO4的 导 带 主 要 由 Ta 5d、In 5s 和 5p 轨 道 贡 献 ,价带主要由 O 2p 轨道贡献,Bi 6s态和 O 2p 态的杂化使 BixIn1-xTaO4固溶体价带位置提高。 另外,通过引入具有半占据d轨道的过渡金属元素也能够调控固溶体的价带位置。例如,Kanhere等57通 过 固 态 反 应 法 制 备 了 (LaFe)x(NaTa)1-xO3固 溶 体光催 化 剂,Fe和 La的 掺 入使 NaTaO3的光吸收范围拓展到 450 nm,其光催化分解水制氢的效率达到 0.81 μmol·h-1·g-1。进一步的理论研究发现,La或者Fe能够在带隙中引入了未占据的杂质能级,虽然能够吸收可将光,但从价带跃迁到杂质态的电子还原能力很弱,不足以还原H+离子产生氢气。然而,当两种元素同时存在时,在(LaFe)x(NaTa)1-xO3固溶体带隙中引入的占据杂质态主要由Fe的3d轨道贡献,这能够抬高价带位置,使材料的带隙减小。 2.3 同时调控导带和价带位置 如果引入的金属离子占据d轨道的能量较高,而未占据的s或p轨道能量较低,固溶体的导带和价带位置能够被同时调控。由于无毒、廉价、高的光吸收效率,以及强的还原能力,ZnS是一种具有 高 活 性 的 产 氢 光 催 化 剂 。 但 其 带 隙 较 宽 (3.7 eV),不具有可见光催化活性,因此其太阳能利用效率很低58。由于 CdS 为窄带隙半导体,其带隙约为 2.4 eV,因而,通过其与 ZnS 形成的固溶体结构是拓展ZnS光响应范围和提高其光催化性能的一种有效方法59-61。Wang 等62在室温下采用无模板的方法制备了 Cd1-xZnxS 固溶体光催化剂,发现当 x= 0.2 且 pH 值 为 7.3 时具 有 最好 的 光催化 产氢性 能 ,在 波 长 大 于 420 nm 可 见 光 照 射 下 的 产 氢 效 率 为458 μmol· h-1。 而 Yu 课 题 组 以 硫 脲 和 Zn(NO3)2· 6H2O 为前驱体,采用水热法制备的 Cd1-xZnxS 固溶体光催化 材料,发现当 x=0.5 时具有最高的光催化产氢效率63,当负载 CdS 量子点时,Zn0.5Cd0.5S 固溶体在 420 nm 光照下的量子效率达到 6.3%64。Dai课题组65基于密度泛函理论的研究表明,Cd1-xZnxS固溶体的价带顶主要由 S 的 3p 态贡献,Cd 4d 态和Zn 3d 态的贡献较 小,其导带底主 要由 Cd 的 5s态和Zn的4s态贡献,另外还有少量S的3s3p态,如图4所示。由于Cd 的4d轨道能量比Zn的3d轨道能量略低,Cd的掺入降低了p-d反键轨道的强度,使价带位置发生了小幅度的下移。但Cd的5s轨道能量比Zn的4s更低,固溶体的导带位置随着Cd 浓度的增加而明显下移。因此,Cd1-xZnxS 固溶体 的 带 隙 随 着 Cd 浓 度 的 增 加 而 减 小66。 半 导 体AgInS2的带 隙 为 1.80 eV,采 用 ZnS 和 AgInS2构建固溶体结构也能够使ZnS的光响应拓展到可见光区 。Kudo 课 题 组67制 备 的 (AgIn)xZn2(1-x)S2固 溶 体 ,带 隙 随 着 x 的 增 大 而 逐 渐 减 小 (如 图 5 所 示)。(AgIn)0.22Zn1.56S2在 420 nm 的可见光照射下分解水的量子效率达到 20%。AgInS2的价带主要由 S 的 3p和Ag的 4d 轨道贡献,而导带主要由 In的5s和 5p轨道贡献。由于 In 的 5s和 5p 轨道比 Zn 的 4s轨道能量 要 低, 固溶 体(AgIn)xZn2(1-x)S2的 导 带位 置随 着 x的增大而降低。另外,S的3p和Ag的4d轨道之间的排斥作用也能够抬高价带位置,AgInS2的引入能够同时改变固溶体的价带和导带位置68。Zhao 等69以 SrCO3、Ag2O、Nb2O5和 P25 为 原 料, 采用 固 态反应法合成的(AgNb)1-x(SrTi)xO3固溶体,能够在可见光照射下分解水产生氢气,通过调节组分可以实现固溶体的带隙在 3.21 到 2.65 eV 范围内调控。纯 SrTiO3具 有 宽 的 带 隙 3.2 eV, 其 价 带 主 要 由 O的 2p 轨 道 贡 献 , 而 导 带 主 要 由 Ti的 3d 轨 道 贡献。由于Ag的4d轨道与O的2p轨道的杂化,使(AgNb)1-x(SrTi)xO3固 溶 体 的 价 带 位 置 抬 高 , 而 Nb 4d 与 Ti 3d 的杂化使导 带 位 置降低。 因 此 ,固溶体的带 隙 明 显减小。Ye 课 题组70采 用 固 态反应法合 成 的 (CaMo)1-x(BiV)xO4固 溶 体 , 带 隙 能 够 随 组分 的 变 化 在 3.64 到 2.34 eV 范 围 内 调 控 。CaMoO4的价带由 O 2p态贡献,而导带由 Mo 4d态贡献。(CaMo)1-x(BiV)xO4固溶体的价带主要由 O 2p和 Bi 6s态贡献,导带主要由 Mo 4d和 V 3d态贡献。由于Bi 6s态和 O 2p态的杂化抬高了价带位置,而 V 3d态和 Mo 4d态的杂化降低了导带位置,固溶体的带隙明显减小。同时改变导带和价带位置,能够在较大的范围内调控固溶体的带隙。 理论 方面 ,Umezawa 等71对于(TiO2)1-x(TaON)x固溶体电子结构的研究发现,TaON 没有在 TiO2带隙中引入局域的杂质态,随着TaON比例的增加,固溶体的带隙逐渐减小。(TiO2)1-x(TaON)x固溶体的价带主要由O和N的2p轨道贡献,而导带主要由Ti的3d轨道和Ta的5d轨道贡献,由于N的2p轨道比 O 的 2p轨道能量更高,(TiO2)1-x(TaON)x固溶体比纯的 TiO2的价带位置更高。而且,阳离子通过π相互作用的d-d成键态也能够降低固溶体的导带位置。因此。制备(TiO2)1-x(TaON)x固溶体的带隙相对于 TiO2显著减小,因此材料能够具有可见光催化活性。Hart等72通过对 GaP-ZnS 体系的理论研究发现固溶体的带隙与原子配位直接相关。GaP的带隙为 2.24 eV,而 ZnS 的带隙为 3.54 eV,12.5%的 GaP引入到 ZnS 中能够使其带隙减小到 1.8 eV。进一步的研究发现Zn―P键和Ga―S键的数目与固溶体的带隙直接相关,Zn的3d轨道和P的3p轨道杂化抬高了价带位置,而Ga的4p轨道与S的3p轨道杂化降低了导带位置。Yang 等73通过实验研 究 的 GaPZnSe 固溶体纳米线,带隙能够在 1.95-2.2 eV 范围内调控,XPS的测量显示固溶体中有新形成的Zn―P和Ga―Se键。 图5 不同组分下(AgIn)xZn2(1-x)S2固溶体的漫反射光谱Fig.5 Diffuse reflection spectra of(AgIn)xZn2(1-x)S2solid solutions for different compositions 固溶体结构不仅能够调控光催化材料的带隙和带边位置,还能够调控其载流子的迁移能力。一般来说,引入元素的轨道比原来的元素更离域,固溶体光生电子或空穴的迁移能力能够得到有效提高。最 近,Hong 课题组74通过固 态反应法制备了 SnNb2-xTaxO6固溶体,在 1.23 V(vsRHE)下的光电流为 9.9 μA·cm-2,相对于纯的 SnNb2O6,固溶体光催化分解水产氢的活性得到显著提高。Sn 5s和 O 2p 轨道之间的杂化抬高了 SnNb2O6的价带位置,使其具有窄的带隙。但Nb的4d轨道比较局域,弱的电子迁移能力使其光催化活性降低。由 于 Ta 的 5d 轨 道 比 Nb 的 4d 轨 道 能 量 更 高 ,SnNb2-xTaxO6固溶体的带隙随着 Ta 浓度的增加而增大。固溶体导带位置的上移增强了光生电子的还原能力,同时,Ta的5d轨道比Nb的4d轨道更加离域,电子的迁移能力也得到提高,这都是提高光催化活性的有利因素。Fan 等75采用一步水热法制备了(Bi2-xYx)Sn2O7固溶体,发现 Y 的混入导致固溶体的光催化活性降低。密度泛函理论的研究表明 ,Y3+替位 Bi3+后, 固 溶体 的 带隙 增大, 而且VBM和CBM的能带结构更加局域,光生载流子迁移能力的减弱使其光催化活性明显降低。 第一性原理计算作为理论研究的有力工具,能够辅助实验发现材料潜在的光催化机理。人们通过计算载流子的有效质量研究它们的迁移性质。电子和空穴的有效质量由导带底和价带顶位置附近的能带拟合得到,这一方程为, 其中ħ为约化普朗克常数,k为波矢,Ek是与波矢k对应的能量。 BiOCl在 降 解 有 机 物 方 面 具 有 与 TiO2相 当 甚至更好的光催化活性,但其带隙较宽,光吸收位于 紫 外 光 区 。 因 此 , 人 们 合 成 了 BiOClxBr1-x76,BiOIxCl1-x77和 BiOBrxI1-x78等固溶 体 增 加 BiOCl的光吸收范围。Zhou 等79基于密度泛函理论研究了系列BiOClxBr1-x、BiOClxI1-x、BiOBrxI1-x固溶体的电子结构和载流子迁移性质。发现随着Br和I浓度的增大,这些体系的空穴有效质量减小非常明显,而电子有效质量几乎保持不变,表明合成这类固溶体结构能够提高其空穴迁移能力。BiOCl的价带主要由O的2p轨道和Cl的3p轨道贡献,而导带主要由 Bi的 6p 轨道贡献,当与 BiOBr或 BiOI形成固溶体结构时,Br和I等阴离子的p轨道和Cl的p轨道之间发生杂化,不仅抬高了价带位置,而且使价带顶变得更加离域,使得空穴迁移能力得到提高。 钙 钛 矿 结 构(ABO3类 型)的 SrTiO3具 有 光 催 化产氢方面的应用前景,但较宽的带隙使其只具有紫外光区的光响应。有研究表明,Cr掺杂能够提高 SrTiO3的可 见 光响 应80,81, 但 Cr3+替位 Ti4+离 子引起的电荷不平衡将在带隙中引入杂质局域态,其作为载流子的复合中心,将降低材料的光催化活性,如果同时引入La元素能够解决电荷不平衡的问题。例如 Harb 和 Umezawa82通过理论研究了钙钛矿结构(SrTiO3)1-x(LaCrO3)x固溶体的电子结构,发现固溶体中 Cr3+替位 Ti4+离子的位置对固溶体的电子结构有很明显的影响。传统固溶体结构中Cr离子的随机分布降低了 Ti―O 键的 pdπ 相互作用,三个 Ti的 t2g态(dxy,dxz,dyz)的宽度减小,不仅增加了电子的有效质量,而且抬高了CBM的位置使发生光跃迁的能量增加,这些因素能够降低该体系的光催化活性。如果合成固溶体材料时能够使Ti和Cr沿[001]方向隔离,将有助于减小电子的有效质量并降低CBM的位置,其总体的光催化活性有望提高。 另外,半导体材料吸收光产生电子空穴对,它们之间的激子效应也会影响其复合。构建固溶体也能够改善激子的解离能力,使载流子更容易发生分离。采用下面的公式估算激子解离能可以判断光生载流子发生解离的难易: 其中RH为氢原子的里德伯常数(13.6 eV),m0为电子质量,εr为宏观静态介电张量,μ为约化的载流子有效质量(1/μ =1/m*e+1/m*h)。 Harb83对 于 固 溶 体(Ta1-xNbx)ON 载 流 子 迁 移 性质的理论研究表明,对于[001]方向,当 Nb 浓度为零时,其空穴有效质量最小,为 0.3m0。当 Nb 浓度为 25%时,该方向的空穴有效质量增加到 0.43m0,而 当 Nb 浓 度 为 50%时 , 空 穴 有 效 质 量 增 加 到0.48m0。在 0%的 Nb 浓度下,对于[100]方向,Nb浓度为零时其电子有效质量同样有最小值,随着Nb 浓 度 的 增 加 , 电 子 有 效 质 量 从 0.33m0增 加 到0.42m0。这些结果表明该固溶体的电子和空穴有效质量均随着Nb浓度的增加而连续增大。另外,(Ta1-xNbx)ON 固溶体沿[001]、[010]和[100]的静态介电常数从 Nb 浓度为零的 20.82、33.71 和 22.51 分别增加到 50%Nb 浓度的 23.65、37.99 和 24.03,表明固溶体方法能够调控材料的介电性质。Dai课题组84从理论 方 面研究了 Ag2ZnSn(S1-xSex)4固 溶 体的电子结构和光催化性质,发现随着Se浓度的增加,固溶体的带隙连续减小,而载流子的迁移能力增强。如表1所示,对静态介电常数和激子束缚能的研究表明,当组分x从0增加到1时,固溶体的平均静态介电常数从 10.19 增加到 13.56,而激子束缚能从 17 meV 减小到 4 meV,说明通过提高固溶体中Se的浓度能够提高该体系的介电性质和激子解离能力,因此,该材料光催化性能有望得到提高。 表1 计算的纯Ag2ZnSnS4、Ag2ZnSnSe4和Ag2ZnSn(S1-xSex)4固溶体的静态介电常数和激子束缚能Table 1 Calculated static dielectric constant and exciton binding energies for Ag2ZnSnS4,Ag2ZnSnSe4,and Ag2ZnSn(S1-xSex)4solid solutions 光催化技术能够直接将太阳能转化为化学能,是解决能源短缺和环境污染问题的理想途径。为了与其它替代技术相竞争,光催化剂需要满足如高效、稳定、无毒、廉价和容易制备等要求。近年来,光催化技术发展迅猛,进展显著,很多国家在这方面投入了巨额的研究资金,但目前仍然存在很多关键问题制约其大规模的应用。为了改善光催化技术,人们发展了固溶体方法,能够实现带隙和带边位置的精确调控,使材料的光响应范围和氧化还原电位之间达到平衡,这是提高材料光催化性能的有效方法之一。另外,固溶体方法还可以调控载流子的分离和迁移性质,这些优点是掺杂等方法所不具备的。 固溶体方法虽然具有诸多优点,但存在的一些不足也影响其光催化性能。由于合适的带隙和离域的价带顶,Zn1-xCdxS 和(GaN)x(ZnO)1-x等含硫或氮元素的固溶体具有较高的光催化活性,但N3-和S2-等离子容易被光生空穴氧化,导致其光稳定性较差。由于助催化剂不仅能够快速转移走光生空穴,减小其对材料的腐蚀,还能作为光催化反应的活性位点和降低反应的能垒,因此通过负载助催化剂可以提高固溶体的光稳定和光催化活 (1) Chen,X.;Shen,S.;Guo,L.;Mao,S.S.Chem.Res.2010,110(11),6503.doi:10.1021/cr1001645 (2)Tong,H.;Ouyang,S.;Bi,Y.;Umezawa,N.;Oshikiri,M.;Ye,J.Adv.Mater.2012,24(2),229.doi:10.1002/adma.201102752 (3) Xiang,Q.;Yu,J.;Jaroniec,M.Chem.Soc.Rev.2012,41(2), 782.doi:10.1039/C1CS15172J (4) Hisatomi,T.;Kubota,J.;Domen,K.Chem.Soc.Rev.2014,43(22),7520.doi:10.1039/C3CS60378D (5)Wang,X.;Maeda,K.;Thomas,A.;Takanabe,K.;Xin,G.; Carlsson,J.M.;Domen,K.;Antonietti,M.Nat.Mater.2009,8(1),76.doi:10.1038/nmat2317 (6)Wang,Z.;Liu,Y.;Huang,B.;Dai,Y.;Lou,Z.;Wang,G.;Qin, X.Phys.Chem.Chem.Phys.2014,16(7),2758.doi:10.1039/ C3CP53817F (7) Yi,Z.;Ye,J.;Kikugawa,N.;Kako,T.;Ouyang,S.;Stuart-Williams,H.;Yang,H.;Cao,J.;Luo,W.;Li,Z.;Liu,Y.; Withers,R.L.Nat.Mater.2010,9(7),559.doi:10.1038/ nmat2780 (8) He,Z.Q.;Tong,L.L.;Zhang,Z.P.;Chen,J.M.;Song,S.Acta Phys.-Chim.Sin.2015,31(12),2341.[何志桥,童丽丽,张志鹏,陈建孟,宋 爽 .物理化学学报,2015,31(12),2341.] doi:10.3866/PKU.WHXB201510151 (9) Chen,B.C.;Shen,Y.;Wei,J.H.;Xiong,R.;Shi,J.Acta Phys.-Chim.Sin.2016,32(6),1371.[陈博才,沈 洋,魏建红,熊 锐,石 兢 .物理化学学报,2016,32(6),1371.] doi:10.3866/PKU.WHXB201603155 (10) Jiang,X.J.;Jia,J.M.;Lu,H.F.;Zhu,Q.L.;Huang,H.F.Acta Phys.-Chim.Sin.2015,31(7),1399.[蒋孝佳,贾建明,卢晗锋,朱秋莲,黄海凤 .物理化学学报,2015,31(7),1399.] doi:10.3866/PKU.WHXB201505191 (11) Lin,C.F.;Chen,X.P.;Chen,S.;Shangguan,W.F.Acta Phys.-Chim.Sin.2015,31(1),153.[林彩芳,陈小平,陈 澍,上官文峰 .物理化学学报,2015,31(1),153.]doi:10.3866/ PKU.WHXB201411175 (12) Peng,Y.;Shang,L.;Cao,Y.;Wang,Q.;Zhao,Y.;Zhou,C.; Zhang,T.Appl.Surf.Sci.2015,358,485.doi:10.1016/j. apsusc.2015.08.025性。另外,固溶体结构中有些元素的溶解度较低,导致其对带隙等性质的可调控范围较小。因此可以考虑通过改变前驱体或合成路径来改善这些问题,例如,Huang 课题组51采用氮化层状双金属氢氧化物合成(GaN)x(ZnO)1-x固溶体,可将 Zn 的浓度显著提高。再者,在固溶体的制备过程中,较高的形成焓能够使固溶体发生相分离或者引入深能级的本征缺陷,也将降低固溶体的光催化活性。因此,通过改变合成条件,合成高结晶度、低缺陷浓度的材料,将使这些问题得到明显改善。 (13) Cao,S.;Zhou,P.;Yu,J.Chin.J.Catal.2014,35(7),989. doi:10.1016/S1872-2067(14)60075-9 (14)Irie,H.;Watanabe,Y.;Hashimoto,K.Chem.Commun.2003,107(23),5483.doi:10.1021/jp030133h (15)Yang,K.;Dai,Y.;Huang,B.J.Phys.Chem.C2007,111(32), 12086.doi:10.1021/jp067491f (16)Yang,K.;Dai,Y.;Huang,B.ChemPhysChem2009,10(13), 2327.doi:10.1002/cphc.200900188 (17) Chen,X.;Liu,L.;Peter,Y.Y.;Mao,S.S.Science2011,331(6018),746.doi:10.1126/science.1200448 (18)Alibabaei,L.;Brennaman,M.K.;Norris,M.R.;Kalanyan,B.; Song,W.;Losego,M.D.;Concepcion,J,J.;Binstead,R.A.; Parsons,G.N.;Meyer,T.J.Proc.Natl.Acad.Sci.2013,110(50),20008.doi:10.1073/pnas.1319628110 (19) Wang,P.;Huang,B.;Qin,X.;Zhang,X.;Dai,Y.;Wei,J.; Whangbo,M.H.Angew.Chem.Int.Edit.2008,47(41),7931. doi:10.1002/anie.200802483 (20)Wang,P.;Huang,B.;Dai,Y.;Whangbo,M.H.Phys.Chem. Chem.Phys.2012,14(28),9813.doi:10.1039/C2CP40823F (21) Ma,X.C.;Dai,Y.;Yu,L.;Huang,B.B.Light:Sci.Appl.2016,5,e16017.doi:10.1038/lsa.2016.17 (22)Yang,H.G.;Sun,C.H.;Qiao,S.Z.;Zou,J.;Liu,G.;Smith,S. C.;Cheng,H.M.;Lu,G.Q.Nature2008,453(7195),638. doi:10.1038/nature06964 (23) Li,B.;Wang,Y.J.Phys.Chem.C2009,114(2),890. doi:10.1021/jp909478q (24)Huang,W.C.;Lyu,L.M.;Yang,Y.C.;Huang,M.H.J.Am. Chem.Soc.2011,134(2),1261.doi:10.1021/ja209662v (25) Wang,H.;Zhang,L.;Chen,Z.;Hu,J.;Li,S.;Wang,Z.;Liu,J.; Wang,X.Chem.Soc.Rev.2014,43(15),5234.doi:10.1039/ c4cs00126e (26) Hong,S.J.;Lee,S.;Jang,J.S.;Lee,J.S.Energy Environ.Sci.2011,4(5),1781.doi:10.1039/C0EE00743A (27) Ran,J.;Zhang,J.;Yu,J.;Jaroniec,M.;Qiao,S.Z.Chem.Soc. Rev.2014,43(22),7787.doi:10.1039/C3CS60425J (28) Liu,L.;Ji,Z.;Zou,W.;Gu,X.;Deng,Y.;Gao,F.;Tang,C.; Dong,L.ACS Catal.2013,3(9),2052.doi:10.1021/cs4002755 (29) Ikeda,T.;Nomoto,T.;Eda,K.;Mizutani,Y.;Kato,H.;Kudo, A.;Onishi,H.J.Phys.Chem.C2008,112(4),1167. doi:10.1021/jp0752264 (30) Klosek,S.;Raftery,D.J.Phys.Chem.B2001,105(14),2815. doi:10.1021/jp004295e (31)Li,Y.;Ding,X.;Zhao,J.;Zhu,Y.;Li,Y.;Deng,W.;Wang,C.Chin.J.Catal.2015,36(12),2170.doi:10.1016/S1872-2067 (15)61018-X (32)Chen,W.;Yang,B.;Yu,Q.;Mao,L.;Fan,Z.;Wang,Q.; Shangguan,W.Appl.Surf.Sci.2015,355,1069.doi:10.1016/j. apsusc.2015.07.204 (33)Alidoust,N.;Toroker,M.C.;Keith,J.A.;Carter,E.A.ChemSusChem2014,7(1),195.doi:10.1002/cssc.201300595 (34) Peng,H.;Ndione,P.F.;Ginley,D.S.;Zakutayev,A.;Lany,S.Phys.Rev.X2015,5(2),021016.doi:10.1103/ PhysRevX.5.021016 (35) Mitchell,R.H.;Chakhmouradian,A.R.J.Solid.State Chem.1999,144(1),81.doi:10.1006/jssc.1998.8121 (36)Wang,Z.;Liu,Y.;Huang,B.;Dai,Y.;Lou,Z.;Wang,G.;Zhang, X.;Qin,X.Phys.Chem.Chem.Phys.2014,16(7),2758. doi:10.1039/c3cp53817f (37) Kato,H.;Kobayashi,H.;Kudo,A.J.Phys.Chem.B2002,106(48),12441.doi:10.1021/jp025974n (38)Ni,L.;Tanabe,M.;Irie,H.Chem.Commun.2013,49(86), 10094.doi:10.1039/C3CC45222K (39) Osorio-Guillen,J.;Lany,S.;Zunger,A.Phys.Rev.Lett.2008,100(3),036601.doi:10.1103/PhysRevLett.100.036601 (40)Zhou,L.;Yu,M.;Yang,J.;Wang,Y.;Yu,C.J.Phys.Chem.C2010,114(44),18812.doi:10.1021/jp107061p (41) Ouyang,S.;Ye,J.J.Am.Chem.Soc.2011,133(20),7757. doi:10.1021/ja110691t. (42) Li,J.;Lu,Q.;Zhang,J.;Zhao,S.Mater.Sci.Semicond.Process2016,49,54.doi:10.1016/j.mssp.2016.03.031 (43)Ma,Z.;Wu,K.;Sun,B.;He,C.J.Mater.Chem.A2015,3(16), 8466.doi:10.1039/C5TA01020A (44)Yuan,Y.;Zhao,Z.;Zheng,J.;Yang,M.;Qiu,L.;Li,Z.;Zou,Z.J.Mater.Chem.2010,20(32),6772.doi:10.1039/C0JM00455C (45) Umezawa,N.;Janotti,A.;Rinke,P.;Chikyow,T.;Van de Walle, C.G.Appl.Phys.Lett.2008,92(4),041104.doi:10.1063/ 1.2835707 (46) Li,Z.;Dong,T.;Zhang,Y.;Wu,L.;Li,J.;Wang,X.;Fu,X.J. Phys.Chem.C2007,111(12),4727.doi:10.1021/jp066671m (47)Li,Z.;Dong,H.;Zhang,Y.;Dong,T.;Wang,X.;Li,J.;Fu,X.J. Phys.Chem.C2008,112(41),16046.doi:10.1021/jp805475a (48) Hu,S.;Zhu,J.;Wu,L.;Wang,X.;Liu,P.;Zhang,Y.;Li,Z.J. Phys.Chem.C2010,115(2),460.doi:10.1021/jp109578g (49) Maeda,K.;Takata,T.;Hara,M.;Saito,N.;Inoue,Y.; Kobayashi,H.;Domen,K.J.Am.Chem.Soc.2005,127(23), 8286.doi:10.1021/ja0518777 (50) Maeda,K.;Xiong,A.;Yoshinaga,T.;Ikeda,T.;Sakamoto,N.; Hisatomi,T.;Takashima,M.;Lu,D.;Kanehara,M.;Setoyama, T.;Teranishi,T.;Domen,K.Angew.Chem.Int.Edit.2010,122(24),4190.doi:10.1002/ange.201001259 (51)Wang,J.;Huang,B.;Wang,Z.;Wang,P.;Cheng,H.;Zheng,Z.; Whangbo,M.H.J.Mater.Chem.2011,21(12),4562. doi:10.1039/C0JM04277C (52) Wang,J.;Yang,P.;Wang,Z.;Huang,B.;Dai,Y.J.Alloy. Compd.2015,652,205.doi:10.1016/j.jallcom.2015.08.190 (53)Wei,W.;Dai,Y.;Yang,K.;Guo,M.;Huang,B.J.Phys.Chem. C2008,112(40),15915.doi:10.1021/jp804146a (54) Sahoo,P.P.;Maggard,P.A.Inorg.Chem.2013,52(8),4443. doi:10.1021/ic302649s (55) Li,G.;Kako,T.;Wang,D.;Zou,Z.;Ye,J.J.Solid State Chem.2007,180(10),2845.doi:10.1016/j.jssc.2007.08.006 (56) Luan,J.;Zou,Z.;Lu,M.;Chen,Y.Mater.Chem.Phys.2006,98(2),434.doi:10.1016/j.matchemphys.2005.09.062 (57) Kanhere,P.;Nisar,J.;Tang,Y.;Pathak,B.;Ahuja,R.;Zheng,J.; Chen,Z.J.Phys.Chem.C2012,116(43),22767.doi:10.1021/ jp307857h (58) Kudo,A.;Sekizawa,M.Catal.Lett.1999,58(4),241. doi:10.1023/A:1019067025917 (59) Zhang,J.;Yu,J.;Jaroniec,M.;Gong,J.R.Nano Lett.2012,12(9),4584.doi:10.1021/nl301831h (60)Li,Q.;Meng,H.;Yu,J.;Xiao,W.;Zheng,Y.;Wang,J.Chem. Eur.J.2014,20(4),1176.doi:10.1002/chem.201303446 (61)Wang,L.;Wang,W.;Shang,M.;Yin,W.;Sun,S.;Zhang,L.Int.J.Hydrog.Energy2010,35(1),19.doi:10.1016/j. ijhydene.2009.10.084 (62)Wang,D.H.;Wang,L.;Xu,A.W.Nanoscale2012,4(6),2046. doi:10.1039/c2nr11972b (63)Li,Q.;Meng,H.;Zhou,P.;Zheng,Y.;Wang,J.;Yu,J.;Gong,J.ACS Catal.2013,3(5),882.doi:10.1021/cs4000975 (64) Yu,J.;Zhang,J.;Jaroniec,M.Green Chem.2010,12(9),1611. doi:10.1039/c0gc00236d (65)Lu,J.;Dai,Y.;Guo,M.;Wei,W.;Ma,Y.;Han,S.;Huang,B.ChemPhysChem2012,13(1),147.doi:10.1002/ cphc.201100527 (66) Wu,J.C.;Zheng,J.;Zacherl,C.L.;Wu,P.;Liu,Z.K.;Xu,R.J. Phys.Chem.C2011,115(40),19741.doi:10.1021/jp204799q (67) Tsuji,I.;Kato,H.;Kobayashi,H.;Kudo,A.J.Am.Chem.Soc.2004,126(41),13406.doi:10.1021/ja048296m (68)Torimoto,T.;Ogawa,S.;Adachi,T.;Kameyama,T.;Okazaki, K.I.;Shibayama,T.;Kudo,A.;Kuwabata,S.Chem.Commun.2010,46(12),2082.doi:10.1039/b924186h (69) Zhao,W.;Ai,Z.;Zhu,X.;Zhang,M.;Shi,Q.;Dai,J.Int.J. Hydrog.Energy2014,39(15),7705.doi:10.1016/j. ijhydene.2014.03.102 (70)Yao,W.;Ye,J.J.Phys.Chem.B2006,110(23),11188. doi:10.1021/jp0608729 (71)Dang,W.;Chen,H.;Umezawa,N.;Zhang,J.Phys.Chem. Chem.Phys.2015,17(27),17980.doi:10.1039/C5CP02110C (72) Hart,J.N.;Allan,N.L.Adv.Mater.2013,25(21),2989. doi:10.1002/adma.201300612 (73) Yang,W.;Liu,B.;Yang,B.;Wang,J.;Sekiguchi,T.;Thorsten, S.;Jiang,X.Adv.Funct.Mater.2015,25(17),2543. doi:10.1002/adfm.201404523 (74) Lee,C.W.;Park,H.K.;Park,S.;Han,H.S.;Seo,S.W.;Song, H.J.;Shin,S.;Kim,D.W.;Hong,K.S.J.Mater.Chem.A2015,3(2),825.doi:10.1039/c4ta05885b (75) Fan,W.;Hu,J.;Huang,J.;Wu,X.;Lin,S.;Huang,C.;Qiu,X.Appl.Surf.Sci.2015,357,2364.doi:10.1016/j. apsusc.2015.09.242 (76) Shenawi-Khalil,S.;Uvarov,V.;Kritsman,Y.;Menes,E.;Popov, I.;Sasson,Y.Catal.Commun.2011,12(12),1136. doi:10.1016/j.catcom.2011.03.014 (77) Wang,W.;Huang,F.;Lin,X.Scr.Mater.2007,56(8),669. doi:10.1016/j.scriptamat.2006.12.023 (78) Jia,Z.;Wang,F.;Xin,F.;Zhang,B.Ind.Eng.Chem.Res.2011,50(11),6688.doi:10.1021/ie102310a (79)Zhang,H.;Liu,L.;Zhou,Z.Phys.Chem.Chem.Phys.2012,14(3),1286.doi:10.1039/c1cp23516h (80)Wang,D.;Ye,J.;Kako,T.;Kimura,T.J.Phys.Chem.B2006,110(32),15824.doi:10.1021/jp062487p (81)Reunchan,P.;Ouyang,S.;Umezawa,N.;Xu,H.;Zhang,Y.;Ye,J.Mater.Chem.A2013,1(13),4221.doi:10.1039/C2TA00450J (82)Chen,H.;Umezawa,N.Phys.Rev.B2014,90(4),045119. doi:10.1103/PhysRevB.90.045119 (83) Harb,M.J.Phys.Chem.C2015,119,4565.doi:10.1021/ jp511878g (84)Jing,T.;Dai,Y.;Ma,X.;Wei,W.;Huang,B.J.Phys.Chem.C2015,119(50),27900.doi:10.1021/acs.jpcc.5b09522 Development of Solid Solution Photocatalytic Materials JING Tao DAI Ying* Traditional semiconductor photocatalysts with a wide band gap can achieve visible light responses through element doping.However,the localized levels introduced by impurities may act as recombination centers of charge carriers,which may lower the photocatalytic activity of the doped materials.The solid solution method can realize precise regulation of the band gap and band edge positions of materials to obtain an optimal balance between their optical absorption and redox potentials.The solid solution method is therefore an effective approach to improve the photocatalytic performance of semiconductor materials.In the present review, considering our recent research,we briefly discuss the latest progress of the solid solution method to tune the band gap and band edge positions of photocatalytic materials as well as examining its influence on carrier separation and migration properties.Finally,challenges and prospects for further development of this method are presented. Photocatalytic material;Doping;Solid solution;Electronic structure;Carrier separation O644 10.3866/PKU.WHXB201610172 Received:August 9,2016;Revised:October 17,2016;Published online:October 17,2016. *Corresponding author.Email:daiy60@sina.com;Tel:+86-531-88365569. The project was supported by the National Key Basic Research Program of China(973)(2013CB632401),National Natural Science Foundation of China(21333006,11374190),and Taishan Scholar Program of Shandong Province,China. 国家重点基础研究发展规划(973)(2013CB632401),国家自然科学基金(21333006,11374190)和山东省泰山学者计划资助项目© Editorial office of Acta Physico-Chimica Sinica

3 对光生载流子的分离和迁移性质的影响


4 结论
(School of Physics,State Key Laboratory of Crystal Materials,Shandong University,Jinan 250100,P.R.China)
