固体酸催化剂结构与催化反应机理的核磁共振研究
2017-03-10李申慧郑安民
李申慧 李 静 郑安民 邓 风
(中国科学院武汉物理与数学研究所,波谱与原子分子物理国家重点实验室,武汉磁共振中心,武汉 430071)
固体酸催化剂结构与催化反应机理的核磁共振研究
李申慧 李 静 郑安民 邓 风*
(中国科学院武汉物理与数学研究所,波谱与原子分子物理国家重点实验室,武汉磁共振中心,武汉 430071)
固体酸催化剂因具有环境友好、高产物选择性和易于分离等优点而广泛应用于现代石油化工领域的各种催化过程。固体核磁共振是一种研究功能材料结构和动力学性质的有力工具。本文将主要介绍固体核磁共振在表征固体酸催化剂的表面酸性以及多相催化反应机理方面的应用。具体来说,可以通过一系列探针分子(如吡啶、丙酮、三烷基氧磷和三甲基磷)的NMR化学位移实验观测值来定量测量酸强度;二维双量子魔角旋转(DQ MAS)固体核磁共振技术能够揭示催化剂上酸中心的空间临近性及协同效应。另外,原位固体核磁共振可以揭示多相催化反应过程中反应物、中间体、反应产物的演化,阐明催化反应机理。
固体核磁共振;酸表征;固体酸催化剂;催化反应机理;主客体相互作用
1 Introduction
Solid acid catalysts such as zeolites,metal oxides and heterpoly acids are important in the catalytic cracking,alkylation,and isomerization reactions in the chemical and petrochemical industry.The widespread application of solid acid catalysts is attributed to their acid-base catalyzed activity1.The catalytic per-formance of solid acid catalysts is mainly attributed to their acidity property and topology structure.In order to better understand the structure-property relationship,the fundamental issues lie in the characterization of acidic property and elucidation of catalytic reaction mechanism.Various well-established spectroscopic techniques including infrared(IR),ultraviolet/visible(UV/Vis), and electron spin resonance(ESR)etc,have been widely utilized to study the solid acid catalysts and related catalytic reactions2. Over the past decades,solid-state NMR spectroscopy has demonstrated a high potential for the characterization of acid sites property and catalytic reactions over solid acid catalysts3-8.In this review,the recent progress on the development and application of solid-state NMR to solid acid catalysis is briefly introduced, which mainly includes(1)quantitative measurements of acid strength of solid acid catalysts by1H,13C and31P chemical shifts of various adsorbed probe molecules(such as pyridine,acetone, trialkylphosphine oxide and trimethylphosphine);(2)spatial proximity and synergetic interaction/effect of different acid sites in solid acid catalysts probed by two-dimensional(2D)1H-1H and27Al-27Al double-quantum magic angle spinning(DQ MAS) NMR spectroscopy;(3)activation and conversion of C1species (methane,CO and methanol)over solid acid catalysts studied by solid-state NMR and density functional theory(DFT)calculations.

LI Shen-Hui,obtained his Ph.D.in 2008 from Wuhan Institute of Physics and Mathematics, Chinese Academy of Sciences.He was appointed as a postdoctoral research fellow at Iowa State University during 2008-2010 and joined WIPM as an associate research fellow since 2010.His current research interests are the development of solid-state NMR methodology for heterogeneous catalysts and materials sciences.

LI Jing,received her B.S.degree from Liaocheng University in 2014.She is currently a joint graduate student under the supervision of Prof.YU Ning-Ya at Hunan Normal University and Dr.LI Shen-Hui at Wuhan Institute of Physics and Mathematics(WIPM), CAS.Her research focuses on solid state NMR studies of host-guest interaction in MOFs.

ZHENG An-Min,obtained his Ph.D.(2005) from Wuhan Institute of Physics and Mathematics,Chinese Academy of Sciences under the supervision of Prof.DENG Feng and has been affiliated with the same institute afterwards.Then he joined Prof.DENG Feng′s group.In 2007,he was a visiting research fellow in Prof.LIU Shang-Bin′s lab at Institute of Atomic and Molecular Sciences,Academia Sinica, Taiwan.In 2012,he was promoted to a professor.His past and current research interest has been focusing on studies of structure and reaction mechanism of the solid acid catalysts by using experimental solid-state NMR and theoretical quantum chemical calculations.

DENG Feng,obtained his Ph.D.(1996)from WIPM,CAS.After his postdoctoral research (1997-1998)at the Department of Chemistry,Texas A&M University(with Prof.James F.Haw),he has worked at WIPM,CAS as a professor(1999 - present).His research interests include solid-state NMR methodology and its application to heterogeneous catalysis.
2 Solid state NMR characterization of acid property of solid acid catalysts
2.1Quantitative measurement of the acid strengthof solid acid catalysts
The acidity of solid acid catalysts dictates their catalytic activity and selectivity during various acid-catalysed reactions.The characterization of the acidic properties is crucial for the design, modification and application of solid acid catalysts.The most important features regarding the acidity characterization focus on the precise determination of the acid type,strength,concentration, distribution and interaction of acid sites.Probe molecules such as, trimethylphosphine(TMP),trimethylphosphine oxide(TMPO), acetone and pyridine were frequently employed to investigate the acidity of solid acid catalysts.13C MAS NMR of adsorbed 2-13C-acetone,31P MAS NMR of adsorbed TMPO and1H MAS NMR of adsorbed pyridine-d5were used to characterize the acidity of various solid acid catalysts9-12.Apart from the experimental observations,theoretical DFT calculations were recently employed to establish the relationship between the1H,13C and31P NMR chemical shifts of the probe molecules and the acid strength of solid acid catalysts13-18.
Proton affinity(PA)or deprotonation energy(DPE)can serve as an indicator for the intrinsic acid strengths of Brønsted acid sites.A smaller PA or DPE corresponds to an easy deprotonation of the acidic proton and thus a stronger Brønsted acid strength.Inthe DFT calculations,proton affinities of Brønsted acid sites varying from 246.7 to 310.8 kcal·mol-1(1 kcal·mol-1=4.187 kJ· mol-1)could be generated covering from weak,medium strong, strong,to super acid.Furthermore,the1H,13C,and31P NMR chemical shifts could be calculated after optimizing the complex structures of the probe molecules adsorbed on the Brønsted acid sites.Thus,the correlations between the Brønsted acid strength and NMR chemical shifts could be established.For pyridine-d5probe molecule,it was found that the1H chemical shift of pyridinium ions decreases linearly with the decrease of PA or the increase of Brønsted acid strength,indicating that the1H chemical shift of adsorbed pyridine-d5can be used as a scale for quantitatively measuring the Brønsted acid strength(Fig.1(a))16.The linear correlation between δ1Hand proton affinity based on the1H-pyridine-d5NMR approach affords quantitative assessment of Brønsted acid strength in solid acid catalysts.Moreover,by taking a PA value of 250 kcal·mol-1as the threshold for superacidity (defined as an acid with strength stronger than 100%H2SO4),an observed1H chemical shift of δ=12.6 could be inferred as the corresponding1H chemical shift threshold for superacidity,which is in well agreement with the experimental value16.Similarly, the31P chemical shift of TMPO probe molecule also exhibits a linear correlation with the proton affinity of Brønsted acid sites (Fig.1(b))17.Different from pyridine-d5,in this case,the31P chemical shift increases linearly with the decrease of PA or the increase of Brønsted acid strength,a larger31P chemical shift corresponds a stronger acid strength,and a31Pchemical shift ofδ= 86 was obtained as the threshold for superacidity17.Extensive characterization of acid site distributions is desirable to understand the detailed reaction mechanism occurring in solid acid catalysts. In order to discern the acid sites located in the internal voids and on the external surfaces of solid acids,Liu et al.19proposed a solidstate31P NMR approach by using trialkylphosphine probe molecules with different sizes,such as trimethylphosphine oxide (TMPO),triethylphosphine oxide(TEPO),tributylphosphine oxide(TBPO),and trioctylphosphine oxides(TOPO).The size of TMPO(kinetic diameter ca 0.55 nm)is smaller than the typical pore aperture of the 10-membered ring(ca 0.60 nm)of zeolite ZSM-5.The small size of TMPO enables it diffuse into the intracrystalline channels and pores of the zeolite.Thus,both the internal and external acid sites are accessible to TMPO.Whereas the size of TBPO(ca 0.82 nm)is too large to penetrate into the channels and can only detect acid sites located on the external surface of the zeolite.Therefore,the concentration of the internal acid sites can be obtained from the difference between those determined from TMPO and TBPO19.Our recent work15further demonstrated that the31P chemical shifts of these trialkylphosphine oxide probe molecules with different sizes could be utilized as a scale for quantitatively measuring the Brønsted acid strength,and a31Pchemical shift ofδ=92-94 corresponds to the chemical shift threshold for superacidity as shown in Fig.2.

Fig.1 Correlations of the calculated1H chemical shift of adsorbed pyridine-d5(a),the31Pchemical shift of adsorbed TMPO(b)and the proton affinity of Brønsted acid sites in solid acid catalysts16,17
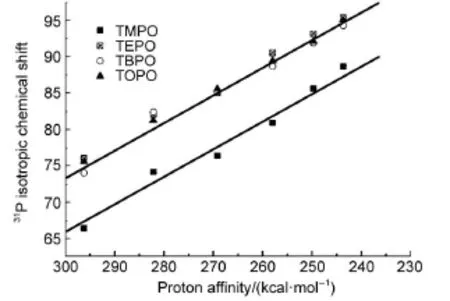
Fig.2 Correlations of the calculated31P chemical shift of adsorbed R3POH+complexes and the proton affinity of Brønsted acid sites in solid acid catalysts15
For 2-13C-acetone probe molecule,it was found that three adsorption conformations(hydrogen-bonded,proton-shared,and ionpair)exist,corresponding to different extents of proton transfer from the Brønsted acid site to the adsorbed acetone14.A correlation of three-broken lines was obtained for the13C chemical shift of acetone versus DPE values.As shown in Fig.3,tri-folded correlation between δ13Cand DPE may be described as followed:Region I(DPE<270 kcal·mol-1)is associated with the formation of ionpair adsorption complexes,corresponding to the presence of strong Brønsted acid sites.Region II(270 kcal·mol-1≤ DPE ≤276 kcal· mol-1)may be related to proton-shared adsorption scheme with medium acidic strength,whereas Region III(DPE >276 kcal·mol-1)belongs to hydrogen-bonded adsorption schemewith relatively weak acidity14.It should be noted that in each region,the13C chemical shift of acetone increases linearly with the decrease of deprotonation energy or the increase of acid strength, which indicates that it can be used as a scale as well for quantitatively measuring the Brønsted acid strength of various solid acid catalysts.A13C chemical shift threshold ofδ=245 was obtained for superacidity in this case(Fig.3),which is in well agreement with the experimental data20.

Fig.3 Correlation of the calculated13C chemical shift of adsorbed 2-13C-acetone with deprotonation energy of Brønsted acid sites in solid acid catalysts14

Fig.4 Correlations of the calculated31P chemical shift against binding energy(which characterizes the Lewis acid strength)of TMP adsorbed on acid sites with B-,Al-,and Ti-Lewis centers13

Fig.5 31PMAS NMR spectra of bare HPMo and TMPO/HPMo samples with varied TMPO loading ranging from 0.5 to 3.3 TMPO/KU21
Trimethylphosphine(TMP)probe molecule can be utilized to discriminate the acid type(BrønstedvsLewis)and quantitatively measure their concentration on solid acid catalysts9.Our recent results indicate that the31P NMR chemical shift of TMP adsorbed on Brønsted acid site is not sensitive to the acid strength,whereas the31P NMR chemical shift of adsorbed TMP on Lewis acid site can be used to determine acid strength of Lewis acids that comprise with the same metallic centers13.Fig.4 shows the computational31P chemical shifts of TMP adsorbed on the Lewis acid systems having different metallic centers,e.g.,BClnF3-n,AlClnF3-n, and TiClnF4-nand their mixed halides.For the TMP-Lewis acid complex,a linear correlation between the calculated31P chemical shifts and corresponding binding energies was observed for the B-, Al-,and Ti-containing Lewis acids,indicating the feasibility of using the31P chemical shift of adsorbed TMP as a scale for Lewis acid strength that comprise with the same metallic Lewis centers13.
Solid-state NMR spectroscopies of adsorbed TMPO and acetone were employed to characterize the acid strength of phosphomolybdic acid(H3PMo12O40,HPMo)21and tungstophosphoric acid(H3PW12O40,HPW)20,22.Fig.5 shows the31PMAS NMR spectra of TMPO adsorbed on the phosphomolybdic acid with different loadings.With the assistance of DFT calculation,resonance peaks with chemical shifts greater thanδ=84 are assigned to the adsorption of one TMPO per Keggin unit(KU)and whereas those with chemical shifts in the range ofδ=80-84 are attributed to (TMPOH+)2/KU.It was conclusive that the acid strengths of Brønsted acidic protons in HPMo,were much stronger than those in typical zeolites,representing solid acid catalysts with superacidic characteristics21.
The above-mentioned acid strength scales have been employed to assess the acidity property of various solid acid catalysts such as AlPW12O4023,mesoporous Nb-W oxides24,flame-derived silica/ alumina25.The correlation between the31P chemical shifts of TMP and the acid strength facilitates the Lewis acidity characterization of mesoporous MoOx/ZrO226,sulfated TiO227,nano particles of zinc oxide28,and niobium oxide29.The established TMPO acid scale was also used to ascertain the acid strength of H-B-ZSM-530and mesoporous carbon-silica solid acid catalysts31.
The influence of acid strength on solid acid-catalyzed protonation of olefins32and alkane activation33was investigated by theoretical calculations.According to the computational results, an increase in acid strength can result in the preferential formation of ion-pair intermediates with greater stability thanπcomplex and alkoxy species intermediates.Moreover,regardless of reactants, intermediates,or transition states,with the increase of acid strength the extent of energy reduction generally follows the order of ion pair > transition state > covalent counterparts(π-complex, alkoxy species).The rule that ionic species are more sensitive than covalent species to acid strength can be used to interpret the effects of acid strength on catalytic activity for other solid-acidcatalyzed reactions,such as alkane activation and isomerization where strongly acidic sites are required34,and Beckmann rear-rangement(BR)reaction over MFI-type zeolites where weakly acidic sites are preferred35.Comprehensive studies have been carried out on the product selectivity and reaction pathway during alkane activation(hydrogen exchange,dehydrogenation,and cracking)and alkene hydrogenation reactions over zeolite catalysts with varied acid characteristics.It is conclusive that catalysts with greater acidic strength are more favorable for enhanced alkane activation during propane cracking,but hydrogen exchange reactions are not sensitive to the acidic strength33.
2.2 Spatial proximity and synergetic effect of acid sites in solid acid catalysts
Zeolite is a kind of most important solid acid catalysts that have been widely used in chemical and petrochemical industry.In zeolites,the Brønsted acid site is a bridging hydroxyl group (SiOHAl)which is associated with the tetrahedral framework Al. In practical applications,zeolites are usually partially dealuminated by a hydrothermal treatment to improve not only their thermal stability but also their catalytic activity.During the dealumination,Al is released from the zeolite framework and the extraframework Al is formed,which acts as the Lewis acid site. The activity enhancement was previously attributed to the Brønsted/Lewis acid synergy effect.However,the existence of the synergy effect is still actively debated and its detailed mechanism is still unclear.
Although one-dimensional(1D)1H and27Al MAS NMR techniques can be utilized to study the structure of both Brønsted and Lewis acid sites,they are unable to provide information about the correlation or interaction between the different acid sites.Fortunately,2D1H-1H DQ MAS NMR spectroscopy36can be applied to investigate the spatial proximity/interaction of different acid sites in various zeolites.The possible synergy effect arising from different active centers,which plays essential role in heterogeneous catalytic reaction processes,can be correlated with their spatial interaction.
Fig.6 shows the 1D1H MAS NMR and 2D1H-1H DQ MAS NMR spectra of dealuminated HY37.As shown in Fig.6(b),two signals atδ =5.0,4.3 are unambiguously assigned to bridging SiOHAl groups in the sodalite and the supercage of HY zeolite, respectively.The peak atδ=2.2 is due to nonacidic SiOH groups. The resonances at δ =2.8,1.0 are attributable to two different types of AlOH hydroxyl groups associated with extra-framework aluminum(EFAL)species that can act as a Lewis acid site37.As shown in Fig.6(c),several types of correlation peaks can be clearly discerned in the1H DQ MAS spectrum of dealuminated HY.The autocorrelation peaks atδ=(4.3,8.6)suggest the spatial proximity of Brønsted acid sites in the supercage.The autocorrelation peak atδ=(2.2,4.4)results from the formation of silanol groups during the dealumination process.Another autocorrelation peak atδ = (2.8,5.6)is due to EFAL species containing more than one hydroxyl group such as Al(OH)3and Al(OH)2+.In addition,the offdiagonal peak pair atδ=(1.0,6.0)and δ=(5.0,6.0)corresponds to the correlation between the extra-framework AlOH group and the bridging hydroxyl group in the sodalite cage,suggesting the spatial proximity between Brønsted and Lewis acid sites.The appearance of another off-diagonal peak pair atδ=(2.8,7.1)and δ=(4.3,7.1)confirms the spatial proximity between the Lewis and Brønsted acid sites in the supercage.Meanwhile,the1H-1H DQ MAS NMR experiments were performed with varying DQ recoupling time to determine the average1H-1H distance between various hydroxyl groups38.Accordingly,the1H-1H distance between a Brønsted acidic proton and an extra-framework AlOH species in the supercage was determined to be 0.43 nm,whereas the average distance between two nearby Brønsted acidic protons in the supercages was measured to be 0.5 nm.The1H-1H DQ MAS NMR spectroscopy was also employed to investigate the spatial proximity of acid sites in H-Y zeolites modified with different calcination,steaming,and acid-leaching treatments,and highly siliceous zeolites,such as H-ZSM-5,H-mordenite,and H-MCM-2239,40.All these findings provide insights into the roles of Lewis acid and its synergy with the Brønsted acid in zeolite-mediated hydrocarbon reactions.

Fig.6 1H MAS NMR spectra of(a)HY,(b)dealuminated HY,(c)2D1H-1H DQ MAS NMR spectra of dealuminated HY37
Apart from the 2D1H-1H DQ MAS NMR,the sensitivity-enhanced 2D27Al-27Al DQ MAS NMR spectroscopy42can also be employed to investigate the spatial interaction among various acid sites in dealuminated zeolites.Fig.7 shows 2D27Al-27Al DQ MAS NMR spectra of parent HY and calcined HY zeolites41.For the parent HY,the autocorrelation peak atδ=(61,122)indicates that four-coordinate framework aluminum(FAL)species are in close proximity to one another(Fig.7(a)).For the HY-500 zeolite as shown in Fig.4(b),the cross peak pair atδ=(61,61)and δ=(0,61)results from the spatial proximity between the four-coordinate FAL and the six-coordinate EFAL,confirming the existence of Brønsted/Lewis acid synergy in the dealuminated HY zeolite.For the HY-600 zeolite,it is revealed that three kinds of aluminum species including four-coordinate FAL,five-coordinate EFAL and six-coordinate EFAL are in close proximity one another(Fig.7 (c))41.For the HY-700,the correlation peak pair atδ=(55,87)and δ =(32,87)is ascribed to the spatial proximity between four-coordinate EFALspecies and five-coordinate EFALspecies(Fig.7 (d)).According to the experimental observations,the detailed spatial correlations among various aluminum species in HY zeolite after dealumination treatment are clearly identified.With the assistance of DFT theoretical calculation,a new dealumination mechanism was proposed and three types of EFAL species,in close proximity to framework aluminum were identified in dealuminated HY zeolites.The spatial proximities of Brønsted and Lewis acid sites in the highly siliceous zeolites,such as H-MOR, H-ZSM-5,and MCM-22 zeolites,were also investigated by the 2D27Al-27Al DQ MAS experiments39,40.It was found that the Brønsted/Lewis acid synergy was present in these highly siliceous zeolites as well.

Fig.7 2D27Al-27Al DQ MAS NMR spectra of(a)parent HY,(b)HY-500,(c)HY-600,and(d)HY-700 zeolites41
As mentioned above,2-13C-acetone is well-established NMR probe molecule for measuring the relative Brønsted acid strengths of solid acids,which was utilized to determine the Brønsted acid strength of dealuminated HY zeolite compared with that of parent HY zeolite37.In the13C NMR spectrum of 2-13C-acetone adsorbed on HY(Fig.8(a)),only one sharp resonance at δ =220 due to unreacted acetone adsorbed on the Brønsted acid site of HY was observed.For the dealuminated HY zeolite as shown in Fig.8(b), with the help of DFT calculations,the signals atδ=228,234 were attributed to acetone adsorbed on two Brønsted acid sites in close proximity to Lewis acid sites with enhanced acid strength, whereas the weak shouldering peak atδ=242 was attributed to acetone adsorbed on Lewis acid sites,suggesting the existence ofBrønsted/Lewis acid synergy in the dealuminated HY zeolite37. Fig.9 summarized the spatial proximities of FAL and EFAL species and Brønsted/Lewis acid synergy in dealuminated H-Y zeolites6.
To measure the Brønsted acid densities in zeolites,Peng et al.43proposed a new approach by using diphenyldiphosphines, Ph2P(CH2)nPPh2,molecules having two basic sites and different chain lengths.2D31P-31P DQ MAS NMR experiments were employed to probe31P-31P inter-nuclear distances and distinguish the nonprotonated,singly protonated,or double protonated diphenyldiphosphines binding sites in the zeolite.Using these techniques,the density and distribution of Brønsted acid sites in HY and H-ZMS-5 zeolites with different Si/Al ratios was comprehensively investigated44.
3 Elucidation of catalytic reaction mechanisms of heterogeneous catalysis
Understanding the reaction mechanisms involved in heterogeneously catalyzed reactions is helpful to refine zeolite catalysts and design new ones.In order to elucidate the catalytic reaction mechanisms,the nature of reactant,intermediate,and product formed on the catalysts during catalytic reaction process needs to be clearly identified45.The initial adsorption states of reactant, reaction intermediates and final product during reaction process can be clearly identified by in situ solid-state MAS NMR spectroscopy3,4,46.Meanwhile,the thermodynamic behavior and kinetic property,strongly related to the catalytic performance,could be theoretically described on the basis of activation barriers and reaction energies derived from the reaction pathway calculations. A combined in situ MAS NMR and theoretical DFT calculation study could provide deep insight into the structure- activity relationship of zeolites in heterogeneously catalyzed reactions
3.1 Methane activation and conversion
Methane,as the cheapest and most abundant natural resource, is the least reactive hydrocarbon molecule due to its strong C―H bond strength.The activation of light alkanes such as methane, ethane and propane at low temperature has attracted intensive experimental and computational studies.The room temperature activation of methane over a Zn modified H-ZSM-5 zeolite catalyst was investigated by solid-state NMR spectroscopy and DFT theoretical calculations47.Fig.10 showed in situ13C MAS NMR spectra of13CH4activation on ZnZSM-5 at the temperature in range of 273 - 333 K.Below 298 K,methane has not been activated and only physically adsorbed methane is visible atδ= -7 (Fig.10(a)).The activation of methane occurs when the temperature reached 298 K,leading to the appearance of a new signal at δ= -20 which was assigned to zinc methyl species(Zn―CH3). The signal atδ=58 was assigned to the surface-bound methoxy species47.The methoxy species is readily transformed into methanol(δ=54)when water is introduced into the sample with pre-activated methane at 298 K(Fig.10(b)).After heating to 523 K(Fig.10(c)),the methanol produced from methane is partially condensed into dimethyl ether(DME,δ=64).Upon consuming all the formed methanol molecules at 573 K,the methanol-to-gasoline(MTG)process finishes with the gasoline components being the major products(Fig.10(d))47.On the basis of the experimental observation,besides zinc methyl species,surface methoxy species attached to zeolite framework was observed at room temperature by in situ MAS NMR.This species plays an intermediate role in the formation of methanol and its further conversion to high-valued hydrocarbons,showing a similar nature to the methoxy species formed from methanol on the acidic zeolites.After a detailed structural characterization,three types of Zn species(isolated Zn2+ion,isolated Zn+ion and Zn+―O-―Zn2+cluster)were identified as the active sites.As shown in Fig.11,at room temperature,the isolated Zn2+ion is responsible for the formation of zinc methyl species via heterolytic dissociation of C―H bond,while the dizinc cluster in open shell is associated with the preferential formation of surface methoxy species through homolytic cleavage of a C―H bond47.The propsed mechanism was also confirmed by DFT calulations.The results may provide valuable insights into the activation mechanism of light alkanes under mild conditions.

Fig.9 Experimentally observed spatial proximities of FAL and EFAL species and Brønsted/Lewis acid synergy in dealuminated H-Y zeolites6
As a clean energy resource and cheap feedstock,selective conversion of methane into useful chemicals attracts general interests from both industrial and academic fields.In situ13C NMRwas employed to monitor the carbonylation of methane with CO to produce acetic acid over a Zn modified ZSM-5 zeolite catalyst48. The13C CP/MAS NMR spectra of products formed from carbonylation reaction of13C-labeled methane with CO on ZnZSM-5 catalyst heated at different temperatures for 1 h were shown in Fig.12.
At 523 K,the signal atδ= - 7 was assigned to unreacted methane and the signal atδ=173 was arising from formate species(Fig.12(a)).Two additional signals were observable atδ= 58,-20,which could be ascribed to surface methoxy species and zinc methyl species,respectively48.The signal of methyl(δ=21) and carbonyl groups(δ=187)indicated the formation of acetic acid resulted from carbonylation reaction at 623 K(see Fig.12(b)). When a trace of molecular oxygen was added,the amount of zinc methyl species(δ= -20)was increased at 523 K while the methoxy species(δ=58)disappeared and the formate(δ=173)and carbonate species(δ=164)was observable(Fig.12(c)).Further increasing the reaction temperature to 623 K resulted in formation of acetic acid(see Fig.12(d)).For comparison,when a trace of H2was adsorbed,the signals of the methoxy species and dimethyl ether(δ=65)and methanol(δ=53)were increased at 523 K, while no zinc methyl group is present(Fig.12(e)).Acetic acid was formed accompanying with the zinc methyl species and ethane (Fig.12(f)).It can be concluded that acetic acid could be generated under a mild reaction condition(573-623 K)through two distinct reaction pathways.In one reaction pathway,methane is activated into methoxy intermediate,which can further interact with CO to generate acetic acid.In another reaction pathway,methane is activated into zinc methyl intermediate that can be consequently transformed into the methyl group of acetic acid with CO2through a typical organometallic reaction48.
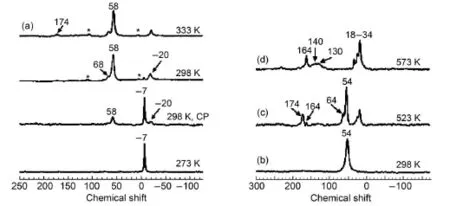
Fig.10 (a)13C NMR spectra of13CH4activation on ZnZSM-5 at specified temperatures and(b)13CH4activation on ZnZSM-5 at 298 K for 2 h and adding water to the sample,(c)heating sample(b)at 523 K for 60 min,(d)heating sample(b)at 573 K for 60 min47

Fig.11 Methane activation and consequent conversion pathway on ZnZSM-5 catalyst47

Fig.12 13C CP/MAS NMR spectra of products formed from coadsorption of CH4and CO on ZnZSM-5 catalyst heated for 1 h48
The direct conversion of methane and carbon dioxide on zincmodified H-ZSM-5 zeolite to produce acetic acid at a low temperature range of 523-773 K was also investigated by solid-state NMR.It was found that the zinc sites efficiently activated CH4to form zinc methyl species(―Zn―CH3),which is further subjected to the CO2insertion to produce surface acetate species(―Zn―OOCCH3)49.For methane activation over Zn-ZSM-5 zeolite,it was found that the oxygen-containing dizinc cluster center in an open shell(Zn+―O-
―Zn2+)was responsible for homolytic cleavage ofthe C―H bond of methane at room temperature47.The reactivity of zinc methyl species over zinc-modified H-ZSM-5 with diverse probe molecules,such as water,methanol,hydrochloride,oxygen, or carbon dioxide could be correlated with that of organozinc compounds in organometallic chemistry50.The alkylation of benzene with methane51and alkylation of benzene with carbon monoxide52to generate toluene over ZnZSM-5 zeolite were also investigated by in situ solid-state NMR.
3.2 Methanol-to-olefins conversion
Methanol-to-olefin(MTO)conversion is one of the most important reactions in C1 chemistry,which provides a chance for producing basic petrochemicals from nonoil resources such as coal and natural gas53.The MTO reaction to produce ethylene and propene over acidic zeolites such as SAPO-34 and ZSM-5 is becoming a promising alternative to the oil route.Mechanistic understanding of the MTO reaction is essential in the rational development of efficient catalysts and catalytic processes and achieving selectivity control of specific olefins54,55.In order to better understand the hydrocarbon-pool(HP)mechanism of methanol-to-olefins(MTO)reaction,the reaction intermediates should be captured and identified.Fig.13 shows13C CP/MAS NMR spectra of the products obtained from the reaction of13C enriched methanol over H-ZSM-5 for 30 min at different temperatures56.At 275 °C,a low methanol conversion was evidenced by the presence of dimethyl ether(δ=60)and methanol(δ=50) (Fig.13(a)).In addition,the formation of pentamethylbenzenium cation was confirmed by the appearance of the signals atδ=189, 205.As the reaction temperature increases to 300-325 °C,the methanol conversion was increased and apart from pentamethylbenzenium ion,1,3-dimethylcyclopentenyl cation,1,2,3-trimethylcyclopentenyl cation,and 1,3,4-trimethyl cyclopentenyl cation were also observable(Fig.13(b,c)).Thus,these carbocations are involved in the MTO reaction over zeolite H-ZSM-5 under working conditions.At higher reaction temperatures,these species were becoming instable reflected by the decrease of their concentration(Fig.13(d,f)).On the basis of the additional GC-MS experimental results and DFT calculations,it was concluded that the formation of propene through a paring mechanism,in which the ring contraction of the pentamethylbenzenium ions leads to the formation of the 1,3,4-trimethylcyclopentenyl cation and propene56.Using similar methods,it was found that the formation of ethene in the methanol to olefins reaction over the H-ZSM-5 zeolite through the aromatics-based paring route in which three types of ethylcyclopentenyl cations were experimentally identified as the key HP intermediates57.Moreover,it was concluded that dimethylbenzene(diMB)and triMB produce ethylcyclopentenyl cations followed by splitting off of ethene,while tetraMB and pentaMB generate propyl-attached intermediates,which eventually produce propene.These results provided new insight into the MBs hydrocarbon pool in MTO chemistry58.The proposed paring routes for the formation of ethene and propene in the MTO reaction over zeolite H-ZSM-5 are illustrated in Fig.14.
The HP mechanism of MTO reaction has also been investigated over chabazite zeolites such as SAPO-34 and H-SSZ-13 by Liu and co-workers54,55,in which both heptamethylbenzenium and pentamethylcyclopentenyl cation intermediates could be identified.The difference for the observation of HP intermediates may be resulted from the pore size difference between chabazite and ZSM-5 zeolites,which play essential roles in formation and stabilization of the carbenium ions.

Fig.13 13C CP/MAS NMR spectra of the products obtained from the reaction of methanol over H-ZSM-5 for 30 min at different temperatures56
3.3 Host-guest interactions in heterogeneous catalysis
The interactions between the reactants and the active sites of zeolites play essential roles in adsorption,desorption and the catalytic reaction process,which would strongly influence the catalytic performance.Investigation of the host-guest interactions may be helpful to explore the catalytic reaction mechanism.The detailed description of host-guest interaction can be extracted by13C-{27Al}symmetry-based rotational-echo saturation-pulse double-resonance(S-RESPDOR)experiments in which the heteronuclear dipolar coupling constant between neighboring13C-27Al spin pair can be quantitatively determined59.
Fig.15 shows13C MAS spectra of 2-13C-acetone adsorbed on dealuminated HY zeolite acquired with and without13C-27Al dipolar dephasing60.The interaction strength between the guestmolecules and the acid sites is directly related to the ratio betweenS(the signal intensity with27Al irradiation)andS0(the signal intensity without27Al irradiation).It can be clearly observable that the signals in range ofδ=228-240 are subject to a strong13C-27Al dipolar dephasing,which could be ascribed to either the hydrogenbond interaction between the carbonyl oxygen of acetone and the Brønsted acidic proton or to acetone directly bound to the Al atom of the Lewis acid site.The relatively smaller dephasing for the carbonyl group(δ=214)can probably be resulted from steric hindrance effects due to a relatively greater size of diacetone alcohol/mesityl oxide.The vinyl groups of mesityl oxide associated with signals in range ofδ=188 - 199 shows smaller13C-27Al dipolar dephasing.The resonance atδ=75 is submitted to an intense13C-27Al dipolar dephasing,which is possibly due to the strong hydrogen-bond interaction between the neighboring OH group in diacetone alcohol and the Brønsted acid site.Therefore, the13C-27Al solid-state NMR technique provides experimental evidence of the interaction models between acetone and Brønsted and Lewis acid sites in dealuminated HY zeolite.By utilizing the13C-{27Al}S-RESPDOR experiment,the13C-27Al internuclear distances between the adsorbed acetone and Brønsted and Lewis acid sites can be quantitatively determined.For acetone adsorbed on Brønsted acid site,the distance is 0.34 nm;for acetone directly bound to Lewis site,the distance is 0.29 nm,which is in good agreement with the DFT predictions.In addition,the spatial interaction between the adsorbed acetone with different active centers(Brønsted and Lewis acids)was also clearly revealed from the 2D27Al-{13C}D-HMQC experiment60.Recently,the27Al-{13C} S-RESPDOR experiment was also empolyed to study the formation oftert-butyl cation confined inside H-ZSM-5 zeolite61.

Fig.14 Proposed paring routes for the formation of ethene and propene

Fig.15 13C MAS NMR spectra for 2-13C-acetone loaded on dealuminated HY zeolite60

Fig.16 13C MAS NMR spectra of trapped products obtained from reactions of methanol over H-ZSM-5 at 300 and 350 °C for 15 min62
It has been also demonstrated that the combination ofin situ13C MAS NMR and27Al-{13C}S-RESPDOR experiment could provide insights into the MTO chemistry62.Fig.16 shows13C RESPDOR NMR spectra of trapped products obtained from reactions of methanol over H-ZSM-5 at 300 and 350 °C for 15 min.At 300 °C, as shown in Fig.16(a),all the signals in the up-field fromδ=0 toδ=60 are subject to different degrees of13C-27Al dipolar dephasing,demonstrating the spatial interaction/proximity between different13C species and framework27Al sites.The signals atδ= 50,60 can be assigned to methanol and dimethyl ether(DME) reactants,respectively,while the signals atδ=59,48 are due to methoxy species typically formed in the MTO reaction and saturated carbon atoms in the rings of cyclopentenyl cations,respectively.With the assistance of two-dimensional1H-13C HETCOR NMR experiment,the signals atδ=17,18 are assigned to the methyl groups of aromatics(mainly pentaMBs and 1,2,3,5-tetraMBs),and the signal atδ=25 to the methyl groups of cyclopentenyl cations and pentamethylbenzenium ion.The27Al-{13C}S-RESPDOR experimental observations suggest the existence of spatial interaction/proximity between trapped HP species and zeolite framework(Brønsted acid site),indicative of the formation of a supramolecular reaction center which was previously proposed by Haw and coworkers to describe the reaction center in MTO process63.By measuring the diploar dephasing fraction(ΔS/S0),it can be expected that MBs interact with theBrønsted acid site(SiOHAl)by formingπ-complex,while the cyclic carbocations interact with the Brønsted base site by forming an ion-pair complex.In combination with12C/13C isotope exchange experiments,it was found that the internuclear spatial interaction/ proximity between the13C nuclei(associated with HP species)and the27Al nuclei(associated with Brønsted acid/base sites)determines the reactivity of the HP species.The closer the HP species to zeolite framework Al,the higher reactivity they possess in the MTO reaction.The formation of the supramolecular reaction center and its influence on the reactivity of the hydrocarbon-pool species provides new insights into the hydrocarbon-pool chemistry in the MTO reaction over zeolite H-ZSM-562.
4 Summary and outlook
Solid acid catalysts have been widely used in industrially important catalytic reactions due to their tunable structural and acidic properties.Solid-state NMR in conjunction with DFT theoretical calculations could be used to characterize the acidity property of solid acid catalysts.The acid strength can be quantitatively measured by the experimentally observed NMR chemical shifts of various probe molecules.The spatial proximity/interaction and synergetic effect of acid sites on zeolites can be manifested from the 2D1H-1H and27Al-27Al DQ MAS NMR spectroscopy.In situsolid-state NMR is able to explore the mechanism of acid-catalyzed reactions by monitoring the evolution of the reactants,intermediates and products,which is helpful to understand the structure-activity relationship of solid acid catalysts.
The application of solid-state NMR to characterize acid sites and elucidate catalytic reaction mechanism on solid acid catalysts is still subject to the relatively lower sensitivity.The enhancement of NMR detection sensitivity can be achieved from the development of versatile NMR pulse techniques as well as high-field spectrometer and high-spinning speed apparatus.Dynamic nuclear polarization(DNP)technique64,65which can enhance NMR signal intensity by 2-3 orders has evolved to become one of the premier methods for structural characterization of heterogeneous catalytic systems,providing in-depth knowledge about catalyst supports, active sites,and their interactions.Para-hydrogen induced polarization(PHIP)technique66,67is favorably considered to be another very promising technique to remarkably enhance NMR signals,which has been successfully employed to clarify a series of industrially important chemical processes68.Additionally,the paramagnetic relaxation enhancement(PRE)solid-state NMR can facilitate the fast acquisition of NMR signals to monitor the heterogeneous catalytic reaction using natural abundance reactants69.
(1) De Vos,D.E.;Dams,M.;Sels,B.F.;Jacobs,P.A.Chem.Rev.2002,102,3615.doi:10.1021/cr010368u
(2) Lysova,A.A.;Koptyug,I.V.Chem.Soc.Rev.2010,39,4585. doi:10.1039/b919540h
(3) Hunger,M.Prog.Nucl.Magn.Reson.Spectrosc.2008,53,105. doi:10.1016/j.pnmrs.2007.08.001
(4)Wang,W.;Hunger,M.Acc.Chem.Res.2008,41,895. doi:10.1021/ar700210f
(5) Li,S.H.;Deng,F.Ann.Rep.NMR Spectrosc.2013,78,1. doi:10.1016/B978-0-12-404716-7.00001-8
(6) Zheng,A.M.;Li,S.H.;Liu,S.B.;Deng,F.Acc.Chem.Res.2016,49,655.doi:10.1021/acs.accounts.6b00007
(7)Zhang,W.P.;Xu,S.T.;Han,X.W.;Bao,X.H.Chem.Soc.Rev.2012,41,192.doi:10.1039/c1cs15009j
(8)Zhang,L.;Ren,Y.H.;Yue,B.;He,H.Y.Chem.Commun.2012,48,2370.doi:10.1039/c2cc16882k
(9) Lunsford,J.H.;Rothwell,W.P.;Shen,W.J.Am.Chem.Soc.1985,107,1540.doi:10.1021/ja00292a015
(10) Biaglow,A.I.;Gorte,R.J.;Kokotailo,G.T.;White,D.J.Catal.1994,148,779.doi:10.1006/jcat.1994.1264
(11) Freude,D.;Hunger,M.;Pfeifer,H.Chem.Phys.Lett.1982,91, 307.doi:10.1016/0009-2614(82)80162-0
(12)Zheng,A.M.;Deng,F.;Liu,S.B.Ann.Rep.NMR Spectrosc.2014,81,47.doi:10.1016/B978-0-12-800185-1.00002-4
(13) Chu,Y.;Yu,Z.;Zheng,A.;Fang,H.;Zhang,H.;Huang,S.J.; Liu,S.B.;Deng,F.J.Phys.Chem.C2011,115,7660. doi:10.1021/jp200811b
(14) Fang,H.;Zheng,A.;Chu,Y.;Deng,F.J.Phys.Chem.C2010,114,12711.doi:10.1021/jp1044749
(15)Zheng,A.;Huang,S.J.;Chen,W.H.;Wu,P.H.;Zhang,H.; Lee,H.K.;de Menorval,L.C.;Deng,F.;Liu,S.B.J.Phys. Chem.A2008,112,7349.doi:10.1021/jp8027319
(16) Zheng,A.;Zhang,H.;Chen,L.;Yue,Y.;Ye,C.;Deng,F.J.Phys.Chem.B2007,111,3085.doi:10.1021/jp067340c
(17) Zheng,A.;Zhang,H.;Lu,X.;Liu,S.B.;Deng,F.J.Phys. Chem.B2008,112,4496.doi:10.1021/jp709739v
(18) Zheng,A.M.;Huang,S.J.;Liu,S.B.;Deng,F.Phys.Chem. Chem.Phys.2011,13,14889.doi:10.1039/c1cp20417c
(19)Zhao,Q.;Chen,W.H.;Huang,S.J.;Wu,Y.C.;Lee,H.K.;Liu, S.B.J.Phys.Chem.B2002,106,4462.doi:10.1021/jp015574k
(20)Yang,J.;Janik,M.J.;Ma,D.;Zheng,A.M.;Zhang,M.J.; Neurock,M.;Davis,R.J.;Ye,C.H.;Deng,F.J.Am.Chem.Soc.2005,127,18274.doi:10.1021/ja055925z
(21)Feng,N.D.;Zheng,A.M.;Huang,S.J.;Zhang,H.L.;Yu,N. Y.;Yang,C.Y.;Liu,S.B.;Deng,F.J.Phys.Chem.C2010,114, 15464.doi:10.1021/jp105683y
(22)Huang,S.J.;Yang,C.Y.;Zheng,A.M.;Feng,N.D.;Yu,N.Y.; Wu,P.H.;Chang,Y.C.;Lin,Y.C.;Deng,F.;Liu,S.B.Chem.-Asian J.2011,6,137.doi:10.1002/asia.201000572
(23) Filek,U.;Bressel,A.;Sulikowski,B.;Hunger,M.J.Phys. Chem.C2008,112,19470.doi:10.1021/jp807947v
(24) Tagusagawa,C.;Takagaki,A.;Iguchi,A.;Takanabe,K.;Kondo, J.N.;Ebitani,K.;Hayashi,S.;Tatsumi,T.;Domen,K.Angew. Chem.Int.Edit.2010,49,1128.doi:10.1002/anie.200904791
(25) Huang,J.;van Vegten,N.;Jiang,Y.J.;Hunger,M.;Baiker,A.Angew.Chem.Int.Edit.2010,49,7776.doi:10.1002/anie.201003391
(26) Xu,J.;Zheng,A.M.;Yang,J.;Su,Y.C.;Wang,J.Q.;Zeng,D. L.;Zhang,M.J.;Ye,C.H.;Deng,F.J.Phys.Chem.B 2006, 110,10662.doi:10.1021/jp0614087
(27)Zhang,H.;Yu,H.;Zheng,A.;Li,S.;Shen,W.;Deng,F. Environ.Sci.Technol.2008,42,5316.doi:10.1021/es800917e
(28) Peng,Y.K.;Ye,L.;Qu,J.;Zhang,L.;Fu,Y.;Teixeira,I.F.; McPherson,I.J.;He,H.;Tsang,S.C.E.J.Am.Chem.Soc. 2016,138,2225.doi:10.1021/jacs.5b12080
(29) Kreissl,H.T.;Nakagawa,K.;Peng,Y.K.;Koito,Y.;Zheng,J.; Tsang,S.C.E.J.Catal.2016,338,329.doi:10.1016/j. jcat.2016.03.007
(30) Wiper,P.V.;Amelse,J.;Mafra,L.J.Catal.2014,316,240. doi:10.1016/j.jcat.2014.05.017
(31) Russo,P.A.;Antunes,M.M.;Neves,P.;Wiper,P.V.;Fazio,E.; Neri,F.;Barreca,F.;Mafra,L.;Pillinger,M.;Pinna,N.;Valente, A.A.Green Chem.2014,16,4292.doi:10.1039/c4gc01037j
(32) Fang,H.J.;Zheng,A.M.;Li,S.H.;Xu,J.;Chen,L.;Deng,F. J.Phys.Chem.C 2010,114,10254.doi:10.1021/jp103247f
(33)Chu,Y.;Han,B.;Fang,H.;Zheng,A.;Deng,F.Microporous Mesoporous Mat.2012,151,241.doi:10.1016/j. micromeso.2011.10.030
(34)Chu,Y.Y.;Han,B.;Zheng,A.M.;Deng,F.J.Phys.Chem.C 2012,116,12687.doi:10.1021/jp302960w
(35)Chu,Y.Y.;Ji,P.;Yi,X.F.;Li,S.H.;Wu,P.;Zheng,A.M.; Deng,F.Catal.Sci.Technol.2015,5,3675.doi:10.1039/ c5cy00619h
(36)Brown,S.P.;Spiess,H.W.Chem.Rev.2001,101,4125. doi:10.1021/cr990132e
(37)Li,S.H.;Zheng,A.M.;Su,Y.C.;Zhang,H.L.;Chen,L.;Yang, J.;Ye,C.H.;Deng,F.J.Am.Chem.Soc.2007,129,11161. doi:10.1021/ja072767y
(38)Li,S.H.;Huang,S.J.;Shen,W.L.;Zhang,H.L.;Fang,H.J.; Zheng,A.M.;Liu,S.B.;Deng,F.J.Phys.Chem.C 2008,112, 14486.doi:10.1021/jp803494n
(39)Yu,Z.W.;Li,S.H.;Wang,Q.;Zheng,A.M.;Jun,X.;Chen,L.; Deng,F.J.Phys.Chem.C 2011,115,22320.doi:10.1021/ jp203923z
(40)Yu,Z.W.;Wang,Q.;Chen,L.;Deng,F.Chin.J.Catal.2012.33, 129.doi:10.1016/s1872-2067(10)60287-2
(41)Yu,Z.W.;Zheng,A.M.;Wang,Q.A.;Chen,L.;Xu,J.; Amoureux,J.P.;Deng,F.Angew.Chem.-Int.Edit.2010,49, 8657.doi:10.1002/anie.201004007
(42)Wang,Q.;Hu,B.;Lafon,O.;Trébosc,J.;Deng,F.;Amoureux, J.P.J.Magn.Reson.2009,200,251.doi:10.1016/j. jmr.2009.07.009
(43)Peng,L.M.;Chupas,P.J.;Grey,C.P.J.Am.Chem.Soc.2004, 126,12254.doi:10.1021/ja0467519
(44)Peng,L.M.;Grey,C.P.Microporous Mesoporous Mat.2008, 116,277.doi:10.1016/j.micromeso.2008.04.014
(45)Hunger,M.Prog.Nucl.Magn.Reson.Spectrosc.2008,53,105. doi:10.1016/j.pnmrs.2007.08.001
(46)Zheng,A.;Huang,S.J.;Wang,Q.;Zhang,H.;Deng,F.;Liu,S. B.Chin.J.Catal.2013,34,436.doi:10.1016/s1872-2067(12) 60528-2
(47)Xu,J.;Zheng,A.M.;Wang,X.M.;Qi,G.D.;Su,J.H.;Du,J. F.;Gan,Z.H.;Wu,J.F.;Wang,W.;Deng,F.Chem.Sci.2012,3, 2932.doi:10.1039/c2sc20434g
(48)Wang,X.;Qi,G.;Xu,J.;Li,B.;Wang,C.;Deng,F.Angew. Chem.Int.Edit.2012,51,3850.doi:10.1002/anie.201108634
(49)Wu,J.F.;Yu,S.M.;Wang,W.D.;Fan,Y.X.;Bai,S.;Zhang,C. W.;Gao,Q.;Huang,J.;Wang,W.J.Am.Chem.Soc.2013,135, 13567.doi:10.1021/ja406978q
(50)Wu,J.F.;Wang,W.D.;Xu,J.;Deng,F.;Wang,W.Chem.-Eur. J.2010,16,14016.doi:10.1002/chem.201002258
(51)Wang,X.M.;Xu,J.;Qi,G.D.;Li,B.J.;Wang,C.;Deng,F. J.Phys.Chem.C 2013,117,4018.doi:10.1021/jp310872a
(52)Wang,X.M.;Xu,J.;Qi,G.D.;Wang,C.;Wang,Q.;Deng,F. Chem.Commun.2014,50,11382.doi:10.1039/c4cc03621b
(53)Tian,P.;Wei,Y.;Ye,M.;Liu,Z.ACS Catal.2015,5,1922. doi:10.1021/acscatal.5b00007
(54)Li,J.Z.;Wei,Y.X.;Chen,J.R.;Tian,P.;Su,X.;Xu,S.T.;Qi, Y.;Wang,Q.Y.;Zhou,Y.;He,Y.L.;Liu,Z.M.J.Am.Chem. Soc.2012,134,836.doi:10.1021/ja209950x
(55)Xu,S.T.;Zheng,A.M.;Wei,Y.X.;Chen,J.R.;Li,J.Z.;Chu, Y.Y.;Zhang,M.Z.;Wang,Q.Y.;Zhou,Y.;Wang,J.B.;Deng, F.;Liu,Z.M.Angew.Chem.Int.Edit.2013,52,11564. doi:10.1002/anie.201303586
(56)Wang,C.;Chu,Y.Y.;Zheng,A.M.;Xu,J.;Wang,Q.;Gao,P.; Qi,G.D.;Gong,Y.J.;Deng,F.Chem.-Eur.J.2014,20,12432. doi:10.1002/chem.201403972
(57)Wang,C.;Yi,X.F.;Xu,J.;Qi,G.D.;Gao,P.;Wang,W.Y.; Chu,Y.Y.;Wang,Q.;Feng,N.D.;Liu,X.L.;Zheng,A.M.; Deng,F.Chem.-Eur.J.2015,21,12061.doi:10.1002/ chem.201501355
(58)Wang,C.;Xu,J.;Qi,G.D.;Gong,Y.J.;Wang,W.Y.;Gao,P.; Wang,Q.;Feng,N.D.;Liu,X.L.;Deng,F.J.Catal.2015,332, 127.doi:10.1016/j.jcat.2015.10.001
(59)Pourpoint,F.;Trebosc,J.;Gauvin,R.M.;Wang,Q.;Lafon,O.; Deng,F.;Amoureux,J.P.ChemPhysChem 2012,13,3605. doi:10.1002/cphc.201200490
(60)Li,S.;Pourpoint,F.;Trebosc,J.;Zhou,L.;Lafon,O.;Shen,M.; Zheng,A.;Wang,Q.;Amoureux,J.P.;Deng,F.J.Phys.Chem. Lett.2014,5,3068.doi:10.1021/jz501389z
(61)Huang,M.;Wang,Q.;Yi,X.;Chu,Y.;Dai,W.;Li,L.;Zheng, A.;Deng,F.Chem.Commun.2016,52,10606.doi:10.1039/ c6cc04943e
(62)Wang,C.;Wang,Q.;Xu,J.;Qi,G.D.;Gao,P.;Wang,W.Y.; Zou,Y.Y.;Feng,N.D.;Liu,X.L.;Deng,F.Angew.Chem.Int. Edit.2016,55,2507.doi:10.1002/anie.201510920
(63)Song,W.G.;Fu,H.;Haw,J.F.J.Am.Chem.Soc.2001,123, 4749.doi:10.1021/ja0041167
(64) Gunther,W.R.;Michaelis,V.K.;Caporini,M.A.;Griffin,R. G.;Roman-Leshkov,Y.J.Am.Chem.Soc.2014,136,6219. doi:10.1021/ja502113d
(65)Ong,T.C.;Liao,W.C.;Mougel,V.;Gajan,D.;Lesage,A.; Emsley,L.;Coperet,C.Angew.Chem.Int.Edit.2016,55,4743. doi:10.1002/anie.201510821
(66)Duckett,S.B.;Mewis,R.E.Acc.Chem.Res.2012,45,1247. doi:10.1021/ar2003094
(67)Giernoth,R.;Heinrich,H.;Adams,N.J.;Deeth,R.J.;Bargon, J.;Brown,J.M.J.Am.Chem.Soc.2000,122,12381. doi:10.1021/ja002516o
(68)Kovtunov,K.V.;Zhivonitko,V.V.;Corma,A.;Koptyug,I.V. J.Phys.Chem.Lett.2010,1,1705.doi:10.1021/jz100391j
(69)Zhou,L.;Li,S.;Su,Y.;Li,B.;Deng,F.Solid State Nucl.Magn. Reson.2015,66-67,29.doi:10.1016/j.ssnmr.2014.12.008
Solid-State NMR Characterization of the Structure and Catalytic Reaction Mechanism of Solid Acid Catalysts
LI Shen-Hui LI Jing ZHENG An-Min DENG Feng*
(State Key Laboratory of Magnetic Resonance and Atomic and Molecular Physics,National Center for Magnetic Resonance in Wuhan,Wuhan Institute of Physics and Mathematics,Chinese Academy of Sciences,Wuhan 430071,P.R.China)
Solid acid catalysts have been widely used in advanced petrochemical processes because of their environmental friendliness,high product selectivity,and easy product separation.Solid-state nuclear magnetic resonance(NMR)spectroscopy is a well-established tool for structure determination and dynamic study of various functional materials.In this review,we focus mainly on our research using solid-state NMR to characterize the acid properties and elucidate the catalytic reaction mechanism of solid acid catalysts.The acid strength of solid acids can be quantitatively measured from the chemical shifts of adsorbed probe molecules such as pyridine,acetone,trialkylphosphine oxides,and trimethylphosphine.The spatial proximity and synergetic effect of various acid sites on solid acid catalysts can be ascertained by two-dimensional(2D)double-quantum magic angle spinning(DQ MAS)NMR spectroscopy.Additionally,in situsolid-state NMR spectroscopy can be used to explore heterogeneous catalytic reaction mechanisms by monitoring the evolution of the reactants, intermediates,and products.
Solid-state NMR;Acidity characterization;Solid acid catalyst;Reaction mechanism; Host-guest interaction
O643
10.3866/PKU.WHXB201611022
Received:September 23,2016;Revised:November 2,2016;Published online:November 2,2016.
*Corresponding author.Email:dengf@wipm.ac.cn;Tel:+86-27-87198820.
The project was supported by the National Natural Science Foundation of China(21210005,21221064,21373265).国家自然科学基金(21210005,21221064,21373265)资助项目© Editorial office of Acta Physico-Chimica Sinica
