氢核磁共振法测定中药茜草中大叶茜草素和羟基茜草素的含量
2017-03-09王志伟闫慧娇王晓陈跃林云良
王志伟,闫慧娇,王晓,陈跃,林云良
(山东省分析测试中心,山东省中药质量控制技术重点实验室,山东 济南250014)
【中药与天然活性产物】
氢核磁共振法测定中药茜草中大叶茜草素和羟基茜草素的含量
王志伟,闫慧娇,王晓,陈跃,林云良*
(山东省分析测试中心,山东省中药质量控制技术重点实验室,山东 济南250014)
综合运用一维和二维核磁共振波谱技术,对茜草提取物中大叶茜草素、羟基茜草素的信号进行指认归属,并建立氢核磁共振法同时测定茜草中大叶茜草素、羟基茜草素含量的方法。在综合考虑茜草提取物的信号分离度及溶解性的基础上,最终选定氘代二甲基亚砜为测试溶剂,以大叶茜草素的烯烃信号(δH5.77, H-2′)和羟基茜草素的H-3信号(δH6.67)为定量峰,以顺丁烯二酸为定量内标。该方法具有较好的稳定性、重复性、精密度和准确性,且测定结果与HPLC-UV方法所得数据基本一致。所建立的氢核磁定量方法操作简单,测试时间短,且定量过程中不需要对照品,可用于中药茜草的质量控制及评价。
茜草;大叶茜草素;羟基茜草素;定量氢核磁共振;质量控制
茜草(RubiacordifoliaL.)作为常用中药,已被《中国药典》收录[1]。茜草含有萘醌类、蒽醌类、三萜类等活性成分[2-3]。其中,大叶茜草素的含量较高,具有抗血小板聚集、保护神经等功效[4-5];而羟基茜草素与茜草质量的优劣存在直接的相关性[6]。因此,大叶茜草素、羟基茜草素的含量常被用于茜草的质量评价。目前,对这两种成分的测定多采用HPLC-UV法,存在耗时、需要标准品等缺点。而羟基茜草素稳定性较差,易被氧化。因此,建立一种快速、可靠的测定茜草中大叶茜草素、羟基茜草素含量的方法具有一定的现实意义。
近年来,定量氢核磁共振法已被广泛应用于中药材、食品、生化样品等复杂体系中活性成分的定量分析[7-15]。2010版《中国药典》已将该方法收录其中。该方法用于定量分析的理论基础是:在适当的实验条件下,定量峰的积分面积与其质子数成正比。当选用合适的内标后,可用于目标化合物的含量测定。该方法简单、快捷,定量分析过程中无需相应化合物的标准品,且对测试样品无损坏。
鉴于目前对茜草提取物中化学成分的相关核磁共振波谱学数据相对不足,对其中主要化学成分的信号归属的基础研究工作相对缺乏等实际问题,本研究在对茜草提取物中大叶茜草素、羟基茜草素结构确定的基础上,采用氢核磁共振建立了茜草中大叶茜草素、羟基茜草素的含量测定新方法,并对18批市售茜草样品进行了分析,测定结果与HPLC-UV所得数据基本一致。
1 材料与仪器
BrukerAvance III 400共振波谱仪,配备Bruker Topspin 3.2工作站(德国布鲁克公司);Varian INOVA-600核磁共振波谱仪(美国瓦里安公司);Agilent 1100型高效液相色谱仪(美国安捷伦公司);Mettler Toledo MX5 电子分析天平(梅特勒-托利多公司)。
大叶茜草素(质量分数99.0%,批号110884-201405)、羟基茜草素(质量分数86.7%,批号111898-201102)标准品购自中国食品药品检定研究院;顺丁烯二酸(质量分数99.0%,批号30318),德国奥格斯堡;DMSO-d6(氘代纯度,摩尔分数99.9%),Cambridge Isotope Laboratories。
茜草(RC-1~RC-18)购自各地药店,共计18批,所有样品均经山东省分析测试中心刘伟副研究员鉴定为茜草科茜草属茜草的干燥根和根茎。
2 方法与结果
2.1 核磁共振(NMR)波谱仪参数设置
2.1.1 BrukerAvance III 400的仪器参数
zg30脉冲序列;弛豫延迟时间(d1) 为6.0 s;采样时间为4.1 s;采样次数为128次;谱宽为11 996 Hz;以Bruker Topspin 3.2软件采集、处理数据。采用手动调整相位和基线,并进行手动积分, 实验过程中以四甲基硅烷信号作为参考确定化学位移标准。每个定量峰重复积分6次,取平均值。
2.1.2 Varian INOVA-600仪器参数
1H NMR的质子共振频率为599.78 MHz,谱宽为7 197 Hz,自由感应衰减信号(free induction decay signal, FID)累加128次。为了对NMR信号归属,采集了样品的氢-氢化学位移相关谱(1H-1H correlated spectroscopy,1H-1H COSY),1H检测的异核多量子相干相关谱(1H detected heteronuclear multiple-quantum coherence,1H-13C HMQC),1H检测的异核多键相关谱(1H detected heteronuclear multiple bond connectivity,1H-13C HMBC)等2D NMR谱。其中,1H-1H COSY的F2(1H)和F1(1H)维的谱宽均为7 634.3 Hz,采样数据点阵t2×t1= 2 290×128,每一条FID累加8次;HMQC的F2(1H)和F1(13C)维的谱宽分别为7 634.3 Hz 和37 700.3 Hz,采样数据点阵t2×t1= 2 290×128,每一条FID累加16次;HMBC的F2(1H)和F1(13C)维的谱宽分别为7 634.3 Hz 和37 700.3 Hz,采样数据点阵t2×t1= 2 290×400,每一条FID累加32次。
2.2 色谱条件
色谱柱为ThermoAcclaimTM120 C18(4.6 mm×250 mm, 5 μm),进样量10 μL,其他条件与《中国药典》一致。
2.3 供试样品的制备
2.3.1 用于茜草提取物中大叶茜草素、羟基茜草素信号归属的样品制备方法
称取4 g经粉碎的茜草药材于锥形瓶中,加入300 mL甲醇,在30 ℃下,超声提取3次,每次15 min,合并后减压回收溶剂至干,加入体积分数为10%的盐酸溶液、氯仿各150 mL,超声2 min,然后在85 ℃下加热回流30 min(重复3次),取氯仿层,合并氯仿层,减压回收溶剂至干,以600 μL DMSO-d6溶解,取500 μL于核磁管中,进行1D NMR及2D NMR测试。
2.3.2 用于定量的样品制备方法
精密称取200 mg经粉碎的茜草药材于锥形瓶中,加入30 mL甲醇,在30 ℃下,超声提取3次,每次15 min,合并后减压回收溶剂至干,加入体积分数为10%的盐酸溶液、氯仿各15 mL,超声2 min,然后在85℃下加热回流30 min(重复3次),取氯仿层,合并氯仿层,减压回收溶剂至干,以600 μL DMSO-d6溶解,精密量取480 μL于核磁管中,并加入20 μL顺丁烯二酸内标溶液(207.16 μg/mL),进行1H NMR测试。精密量取剩余样品溶液100 μL,以900 μL甲醇稀释至1 000 μL,经0.45 μm微孔滤膜滤过后,用于HPLC-UV 分析。用于样品测试及方法学考察的顺丁烯二酸内标溶液的配制方法:精密称取5.179 mg顺丁烯二酸,以1 mL DMSO-d6溶解。
2.4 定量化合物的NMR数据归属
首先,我们通过2D NMR对茜草提取物中大叶茜草素、羟基茜草素1D NMR数据进行了归属。在COSY谱中,在δH5.77的双重峰(d, 8.4 Hz)与δH6.85的双重峰(d, 9.0 Hz)存在相关;在HMQC谱中,该质子与δC130.3处的叔碳信号相关;在HMBC谱中,在δH1.40处的甲基氢信号与δC130.3和δC75.3相关,通过以上信息并结合其他信号归属,确定δH5.77为大叶茜草素中H-2′的烯氢信号。在HMQC谱中,δH6.67处的单峰与δC110.4叔碳信号存在相关;在HMBC谱中,该氢信号与δC160.8,δC157.6,δC149.7,δC105.6处的季碳信号相关,以上信息表明δH6.67应为羟基茜草素的H-3信号。详细的大叶茜草素、羟基茜草素的化学位移数据见表1,结构见图1。这是首次对茜草提取物中大叶茜草素、羟基茜草素的NMR数据进行指认归属。
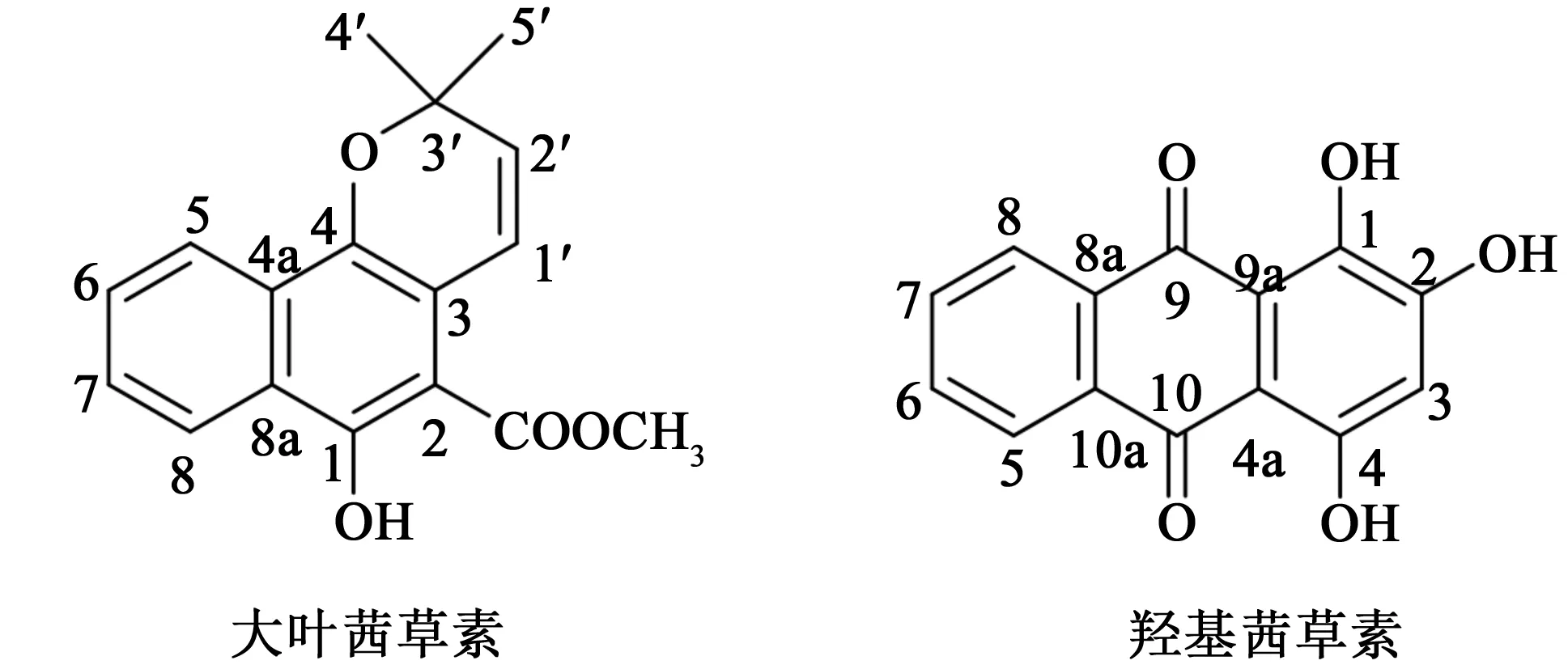
图1 大叶茜草素、羟基茜草素的结构式Fig.1 Structures of mollugin and purpurin

化合物位置δH(J/Hz)δCHMQC1H⁃1HCOSYHMBC大叶茜草素1-151.9--H⁃82-106.0--H⁃1′3-112.8--H⁃2′4-141.2--H⁃1′,H⁃558.04(d,8.0)121.9+H⁃6H⁃767.61(t,6.6)129.4+H⁃5,H⁃7H⁃877.53(t,7.2)127.0+H⁃6,H⁃8H⁃588.19(d,8.4)123.8+H⁃7H⁃64a-127.8--H⁃6,H⁃88a-125.1--H⁃5,H⁃71′6.85(d,9.0)121.6+H⁃2′-2′5.77(d,8.4)130.3+H⁃1′H⁃4′/5′3′-75.3--H⁃4′/5′4′/5′1.40(s)27.0+-H⁃2′⁃COOCH33.91(s)53.0+--⁃COOCH3-170.6--⁃COOCH3羟基茜草素1-149.7--H⁃32-157.6--H⁃336.67(s)110.4+--4-160.8--H⁃358.08(d,7.8)126.9+H⁃6H⁃767.86(m)134.6+H⁃5H⁃577.81(t,7.8)134.3+H⁃8H⁃888.13(d,7.2)127.0+H⁃7H⁃69-184.0--H⁃810-182.6--H⁃54a-105.6--H⁃310a-133.1--H⁃88a-133.0--H⁃79a-113.0---
2.5 定量峰的确定
以DMSO-d6为1H NMR测试溶剂时,顺丁烯二酸的信号峰δH6.28,大叶茜草素的烯烃信号δH5.77 (H-2′),羟基茜草素的H-3信号δH6.67与茜草提取物中其他信号具有较好的分离度,可用于大叶茜草素、羟基茜草素的1H NMR定量。大叶茜草素、羟基茜草素标准品及茜草提取物的1H NMR谱见图2。

1 大叶茜草素;2 羟基茜草素;IS 顺丁烯二酸。图2 对照品及茜草样品 1H NMR谱图Fig.2 The 1H NMR spectra of reference compounds and Rubiae Radix et Rhizoma extract
2.6 纵向弛豫时间T1的测定
称取大叶茜草素0.380 mg、羟基茜草素0.260 mg、顺丁烯二酸0.654 mg, 分别置于核磁管中,加入0.5 mL DMSO-d6溶解,用于1H NMR测试。纵向弛豫时间T1采用质子反转-恢复T1实验方法进行测定,并用Bruker的T1计算程序计算。设定脉冲弛豫延迟时间范围为1 ~30 s。经测定,用于大叶茜草素、羟基茜草素定量的信号的T1值分别为1.5 s,2.0 s,顺丁烯二酸烯烃信号的T1值为2.5 s。
2.7d1值的优化
首先我们测定了大叶茜草素、羟基茜草素、顺丁烯二酸定量峰的纵向弛豫时间T1值。采用90°脉冲测定样品时,为满足d1≥5T1max的要求,d1值最小为12.5 s,这种情况下样品测试时间较长。在确保准确性的前提下,本文采用30°脉冲,考察不同d1值(1~15 s,1 s间隔)对大叶茜草素、顺丁烯二酸定量信号积分面积的影响。结果表明,与90°脉冲,d1值为15 s时测定的积分值(积分面积完全反映该信号的质子数)相比较,采用30°脉冲,d1值为6 s时可以达到99.1%的“回收率”,大大缩短了样品的测定时间。因此,经优化后采用的仪器测定参数为30°脉冲,d1值6 s,128次扫描,采样时间为4.1 s。
2.8 准确性考察
精密称取大叶茜草素4.978 mg,羟基茜草素3.575 mg,以1 000 μL DMSO-d6溶解后,分别精密量取400 、200 、100 、50 、25 、12.5 、6.3 μL置于核磁管中,各加入20 μL顺丁烯二酸内标溶液,分别加入80 、280 、380 、430 、455 、467.5 、473.7 μL DMSO-d6后进行1H NMR测定。以理论浓度为横坐标,以实测浓度为纵坐标,进行回归分析。信噪比3和10时分别为最低检测限和最低定量限。如图3所示,大叶茜草素公式为y=0.967x+0.025,羟基茜草素公式为y=0.967x+0.009,实测值与理论值基本一致,相关系数分别为0.999 9, 1.000 0,结果表明该方法具有较好的准确性。大叶茜草素最低检测限和最低定量限分别为7.59 μg/mL、22.76 μg/mL;羟基茜草素最低检测限和最低定量限分别为10.52 μg/mL、31.56 μg/mL。

图3 大叶茜草素、羟基茜草素标准溶液理论浓度与测定浓度回归分析图Fig.3 Relationships between mollugin and purpurin concentrations determined by quantitative 1H NMR and the theoretical concentrations
2.9 精密度试验
取含大叶茜草素3.982 mg/mL、羟基茜草素2.846 mg/mL、顺丁烯二酸207.16 μg/mL的标准溶液连续测试6次,连续测试3 d,分别考察仪器日内和日间精密度,结果显示以积分面积的相对标准偏差计大叶茜草素、羟基茜草素的日内精密度分别为0.8%,0.7%,日间精密度分别为0.8%,1.2%,说明仪器精密度良好。
2.10 重复性试验
取同一样品6份(RC-18),按照2.3.2项方法平行制备供试品溶液,按2.1.1项设置NMR仪器参数。大叶茜草素、羟基茜草素积分面积的相对标准偏差分别为3.5%,1.7%,表明重复性良好。
2.11 稳定性试验
取含大叶茜草素、羟基茜草素、顺丁烯二酸分别为3.982 、2.846 、207.16 mg/mL的标准溶液,每2 h进行一次1H NMR测试,连续测定24 h。经测定,大叶茜草素、羟基茜草素积分面积的相对标准偏差分别为1.4%,1.6%,表明供试样品24 h内稳定性良好。
2.12 加样回收率试验
精密称取9份已知大叶茜草素含量的RC-18样品200 mg,分为3组,每组分别精密加入相当于原质量分数50%、100%、150%的大叶茜草素标准品。按照2.3.2项的方法制备供试样品,计算加样回收率。结果表明,大叶茜草素平均回收率为94.7%~103.2%,相对标准偏差均小于3.9%。
2.13 样品分析
利用建立的1H NMR定量方法,分析了18批各地药店市售茜草药材中大叶茜草素、羟基茜草素的含量,并将本方法的结果与HPLC-UV测定数据进行了比较,如表2所示,两种方法所得结果基本一致。
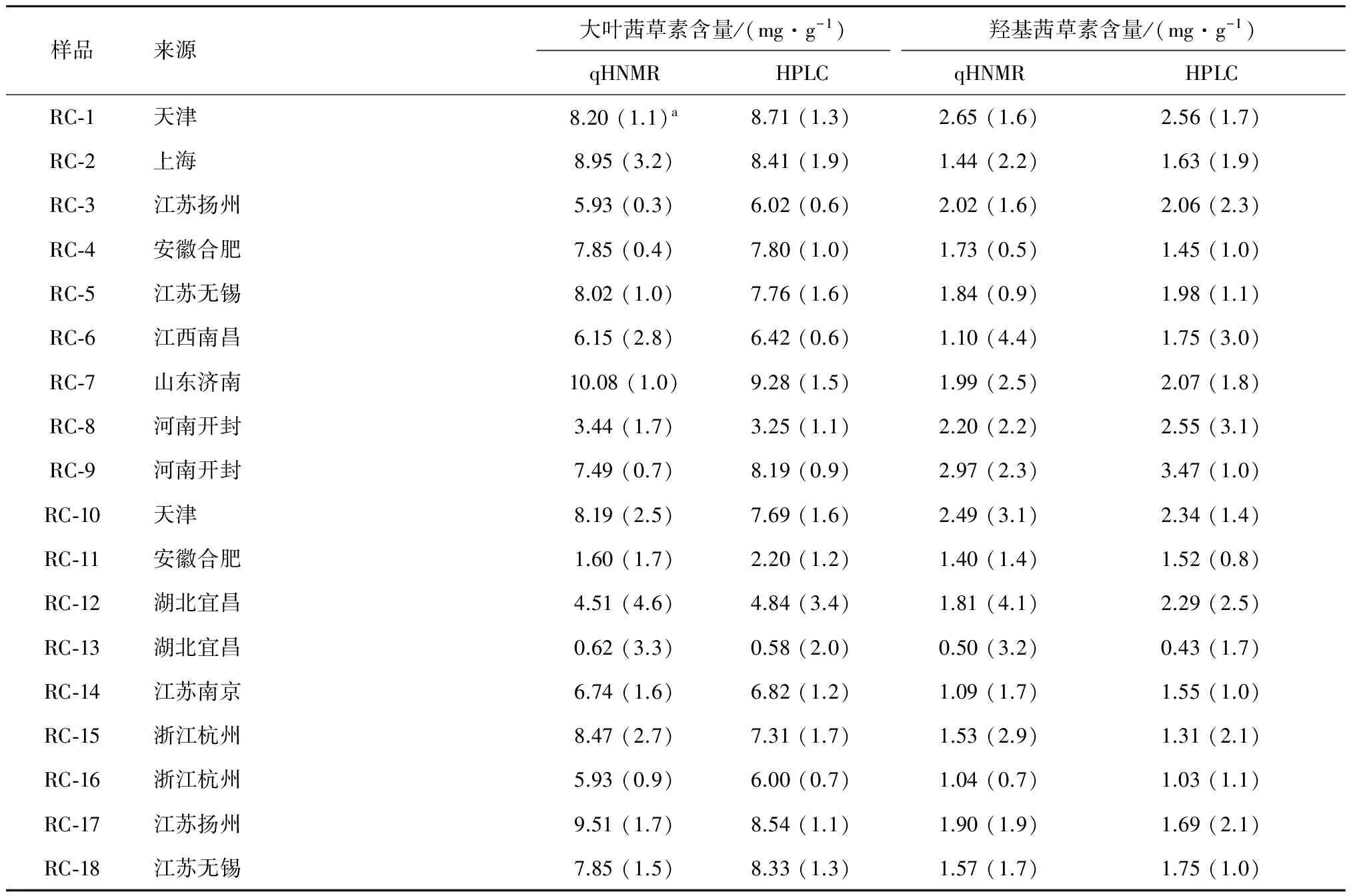
表2 茜草药材中大叶茜草素、羟基茜草素含量(mg/g)
注:a为相对标准偏差 (n= 3)。
3 讨论
结果表明,各地药店市售茜草药材中大叶茜草素(0.62 ~10.08 mg/g)的含量较高,而羟基茜草素的含量较低(0.50 ~2.97 mg/g),各样品间两化合物的含量相差较大,药材质量参差不齐。
本方法能同时分析中药茜草中大叶茜草素、羟基茜草素的含量,分析时间较短,具有较好的准确性和精密度,定量过程中通过选择顺丁烯二酸作为内标,避免使用相应的标准品,可用于中药茜草的质量评价,对建立其他中药材的氢核磁共振定量方法具有一定的指导意义。
[1]国家药典委员会.中华人民共和国药典2015年版一部 [M]. 北京:中国医药科技出版社,2015: 234.
[2]ITOKAWA H, MIHARA K, TAKEYA K. Studies on a novel anthraquinone and its glycosides isolated from Rubiae Radix et Rhizoma andR.akane[J]. Chem Pharm Bull, 1983, 31(7): 2353-2358.
[3]ITOKAWA H, QIAO Y F, TAKEYA K, et al. New triterpenoids from Rubiae Radix et Rhizoma var.pratensis(Rubiaceae) [J]. Chem Pharm Bull, 1989, 37(6): 1670-1672.
[4]CHUNG M I, JOU S J, CHENG T H, et al. Antiplatelet constituents of formosanRubiaakane[J].J Nat Prod, 1994, 57(2): 313-316.
[5]JEONG G S, LEE D S, KIM D C, et al.Neuroprotective and anti-inflammatory effects of mollugin via up-regulation of heme oxygenase-1 in mouse hippocampal and microglial cells[J]. Eur J Pharmacol, 2011, 654(3): 226-234.
[6]陈星, 王侃,单鸣秋,等.UPLC测定茜草炭中4种醌类成分的含量[J].中国中药杂志, 2012, 37(19): 2922-2925.
[7]TANAKA R, NITTA A, NAGATSU A. Application of a quantitative1H-NMR method for the determination of amygdalin in Persicae semen, Armeniacae semen, and Mumefructus[J].J Nat Med, 2014, 68(1): 225-230.
[8]HOHMANN M, FELBINGER C, CHRISTOPH N, et al. Quantification of taurine in energy drinks using1H NMR[J]. J Pharm Biomed Anal, 2014, 93: 156-160.
[9]PIERI V, BELANCIC A, MORALES S, et al.Identification and quantification of major steviol glycosides inSteviarebaudianapurified extracts by1H NMR spectroscopy[J]. J Agric Food Chem, 2011, 59(9): 4378-4384.
[10]LI C Y, XU H X, HAN Q B, et al.Quality assessment of Radix Codonopsis by quantitative nuclear magnetic resonance[J].Journal of Chromatography A, 2009, 1216(11): 2124-2129.
[11]WANG Z W, WANG J S, YANG M H, et al. Developmental changes in the composition of five anthraquinones fromRheumpalmatumas quantified by1H-NMR[J]. Phytochem Anal, 2013, 24(4): 329-335.
[12]HOLZGRABE U.Quantitative NMR spectroscopy in pharmaceutical applications[J]. Prog Nucl Magn Reson Spectrosc, 2010, 57(2): 229-240.
[13]NAPOLITANO J G, GÖDECKE T, RODRGUEZ-BRASCO M F, et al.The tandem of full spin analysis and qHNMR for the quality control of botanicals exemplified withGinkgobiloba[J].J Nat Prod, 2012, 75(2): 238-248.
[14]PAULI G F, GÖDECKE T, JAKI B U, et al. Quantitative1H NMR: Development and potential of an analytical method: An update[J]. J Nat Prod, 2012, 75 (4): 834-851.
[15]BURTON I W, QUILLIAM M A, WALTER J A. Quantitative1H NMR with external standards: Use in preparation of calibration solutions for algal toxins and other natural products[J]. Anal Chem, 2005, 77(10): 3123-3131.
Quantitative determination of mollugin and purpurin from traditional Chinese medicine Rubiae Radix et Rhizoma by1H NMR
WANG Zhi-Wei, YAN Hui-Jiao, WANG Xiao, CHEN Yue, LIN Yun-Liang*
(Shandong Provincial Key Laboratory for TCM Quality Control Technology, Shandong Analysis and Test Center,Shandong Academy of Sciences, Jinan, 250014)
∶ The1H chemical-shift values of mollugin and purpurin in extract from Rubiae Radix et Rhizoma were assigned on the basis of 1D and 2D NMR, and a specific, simple, sensitive1H NMR method has been developed and validated for the simultaneous quantitative determination of mollugin in extract from Rubiae Radix et Rhizoma When the solvent effects on the resolution of target signals and the solubility of the crude extracts were taken into account, dimethyl sulfoxide-d6was selected as an optimal1H NMR solvent. Quantitative determination was carried out by using the signal of the olefinic proton of mollugin (δH5.77, H-2′) and H-3 of purpurin (δH6.67), which were not interfere with other signals. Maleic acid was selected as internal standard because of its stable physical and chemical properties and having simple resonance signal. The improved method possessed good stability, repeatability, precision and accuracy for the quantification of mollugin and purpurin and the determination results agreed with the data obtained by HPLC-UV method. Due to its simple operation, short test time and without any reference compound, the proposed quantitative1H NMR method can alternatively be used for the quality control of Rubiae Radix et Rhizoma.
∶Rubiae Radix et Rhizoma; mollugin; purpurin; quantitative1H NMR; quality control
10.3976/j.issn.1002-4026.2017.01.001
2016-05-09
山东省科学院青年科学基金(2014QN005);山东省自然科学基金三院联合基金(ZR2015YL012)
王志伟(1983—),男,博士,研究方向为中药效物质基础及质量控制,E-mail:wzwcpu@163.com
*通信作者,林云良。 Tel:0531-82605343;E-mail:wodekafei@126.com
R284.1
A
1002-4026(2017)02-0001-07
