基于梯度提升决策树的肽碎片离子强度建模
2017-03-09张龙波王晓丹
怀 浩,刘 学,张龙波,王晓丹
(山东理工大学 计算机科学与技术学院,山东 淄博 255049)
基于梯度提升决策树的肽碎片离子强度建模
怀 浩,刘 学,张龙波,王晓丹
(山东理工大学 计算机科学与技术学院,山东 淄博 255049)
为找到对蛋白质鉴定算法影响较大的肽碎片离子特征,以提高鉴定结果的正确率,在碎片离子特征与强度信息的基础上进行建模,构建预测模型. 实验首先使用pFind对串联质谱数据鉴定,将鉴定结果过滤出需要的肽序列;然后计算出离子质荷比与离子特征值,通过匹配离子的质荷比获取离子强度信息;使用强度信息与离子特征值构建libsvm格式文件,使用XGBoost构建预测模型,其中使用了梯度提升决策树算法;最后使用构建完成的预测模型对蛋白质产生的肽序列做离子强度理论预测.实验结果表明模型所预测的肽序列离子强度与实验离子强度有着较高的相似度,同时分析预测模型可以从预测树中发现肽序列碎裂的规律,提取肽碎片离子中对强度值影响较大的离子特征.
串联质谱;肽碎片离子强度;梯度提升决策树;建模
基于串联质谱的识别算法—肽谱匹配(peptidespectrummatching,PSM)是一种高健壮的得分算法,也是肽碎片离子精准建模的主要依据.通过串联质谱数据识别肽,大多数软件依靠于对比实验图谱和理论图谱,因此提高理论图谱的准确度可以提高肽识别的准确率[1].肽碎片离子的强度信息可以用来分析各种离子特征在裂解途径上对肽碎裂的影响.分析这些信息可以帮助开发更多可靠的肽和蛋白质识别算法,同时重要的特征信息可以被用来预测质谱强度信息,并将这些信息用于对肽和蛋白质鉴定的算法中,提高理论图谱的准确性.针对于碎片离子强度预测,其首要工作是构建模型,文献[2-6]根据不同的碎裂离子特征构建出了不同的模型,实验在此基础上,选取文献中对离子强度影响较大的离子特征,以提高算法对离子强度预测的准确度,同时结合离子强度,使用机器学习的方法,在碎片离子强度信息的基础上进行建模,构建预测模型.
1 算法模型
1.1XGBoost
XGBoost是一个设计高效,灵活并且可移植的最优分布式决策梯度提升库.它实现了梯度提升框架下的机器学习算法[7-8].XGBoost提供平行提升树,它可以实现快速和准确的解决许多数据科学问题.相同的代码能够在各大分布式环境(Hadoop,SGE,MPI)中运行,并可以解决超过十亿数据的问题.XGBoost的特点有:速度快,可移植,效果好,功能多.
XGBoost使用梯度提升决策树(GradientBoostingDecisionTree,GBDT)算法.1999年由JeromeFriedman提出,将GBDT模型应用于ctr预估.GBDT是一个加性回归模型,它通过Boosting的迭代构造一组弱学习器.该算法由多棵决策树组成,最终结果将多棵树的结果累加起来.决策树模型非常适合于学习不同规则集的肽碎片[9]裂解规律.Boosting是对一组数据,通过构建多个弱分类模型,然后下一次分类会将在上一次分错的数据权重提高一点再进行新的分类模型构建,这样最终得到的分类器在测试数据与训练数据上都可以得到比较好的效果.
沿着梯度方向,构造一系列的弱分类器函数,并以一定权重组合起来,形成最终决策的强分类器.
(1)
其中的ω是权重,φ是弱分类器(回归器)的集合,公式可以理解为一个加法模型.
1.2 评价指标
使用相似度[2]作为模型的评价指标.用相似度公式计算预测强度与理论强度个体间的相似程度,数值越大,说明个体间相似度越大,预测的结果越准确.式(2) 和式 (3) 分别对应图谱相似度的计算公式以及简化公式:
(2)
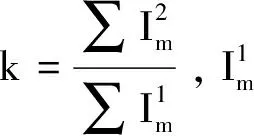
(3)
2 实验与分析
本实验中串联质谱数据的鉴定使用的是数据库搜索方法,它是当前高通量蛋白质的主要鉴定方法,该方法使用谱——谱比对的方式,避免了蛋白质鉴定中理论图谱预测的难点.鉴定过程中所使用到的质谱数据的离子强度特征信息是鉴定蛋白质的关键因素.串联质谱法的步骤为:蛋白质经过酶切后通过一级质谱分析器,从一级质谱中选取部分强度较高的肽段进入二级质谱分析器,分析出含有肽段序列信息的串联质谱数据,通过数据库搜索方法鉴定出肽序列,进而推断出蛋白质[1].串联质谱数据由于具有独立性和可解析性,因此可以通过相关软件(如Xcalibur)观测图谱.
通过实验收集和计算的大量质谱数据,不仅可以用于蛋白质的鉴定,同样可以使用于机器学习的分析方法.通过机器学习不需要明确了解背后建模的基本机理,相反,可以直接利用机器学习来发现数据中隐藏的关联模式数据中的相关性,并利用它们得出有用的结论.
实验操作系统平台为WindowsServer2008R2Enterprise,硬件平台为2.00GHz的IntelXeonE5-2620处理器,32GB内存.质谱数据的鉴定使用pFind软件,鉴定结果数据的处理代码使用python语言编写,模型构建工具在python下使用XGBoost工具库.
2.1 实验步骤
实验首先对串联质谱数据进行预处理,之后使用pFind对串联质谱数据鉴定.根据设定条件过滤出所需要的肽序列,通过公式计算出离子质荷比与离子特征值,通过质荷比进行肽谱匹配来获取离子强度值.然后使用强度信息与离子特征值构建libsvm数据集,使用XGBoost构建预测模型.最后使用构建完成的预测模型对蛋白质产生的肽序列做离子强度理论预测.
2.1.1 数据预处理
实验采用Hela与Yeast两个串联质谱数据集,文件大小分别为4.36GB与4.21GB.对于原始的raw格式数据源使用msconvert软件将其转换为mgf格式数据源,再将其使用pFind进行蛋白质的鉴定.实验时分别对Hela数据集与Yeast数据集进行独立建模实验.
串联质谱数据可以看作是一个两维数组,第一维表示离子质荷比,第二维表示质荷比所对应的离子峰强度,由于在目前的蛋白质鉴定系统中,质荷比信息的应用较离子强度更加广泛,因此如何利用离子强度信息是一个值得深入研究的重要课题,本实验正是在此基础上利用离子强度建模与预测.
实验选用pFind作为鉴定软件,使用Trypsin酶切,固定修饰:Carbamidomethyl(C),可变修饰:Acetyl(N-term)和Oxidation(M),母离子质量误差:±0.007Da,碎片离子质量误差:±0.02Da,最大2个遗漏酶切位点.
实验使用假阳性率(falsediscoveryrate,FDR)[10],将其阈值设置为0.1%,来确保鉴定出的肽集合有较高的可信度,因为即使是所谓“纯”的质谱,也可能含有未赋值的峰值[11].
由表1可知不含氧化修饰的序列可以到达70%以上,母离子电荷为2的序列可占总序列的65%以上,而且序列中存在大量重复肽序列,对重复的肽序列处理方式为保留得分最大的一个,因此鉴定结束后,通过pFind导出结果文件,方便计算碎片离子属性值,对实验结果进行处理: (1)去除肽序列有重复的以及反向数据库的肽序列数据;(2)去除含有氧化修饰的数据; (3)只取母离子电荷为2的数据.
2.1.2libsvm格式文件构建
先取出10%的数据留作模型测试数据(measuredata),将剩余数据分为10份,使用10折交叉验证法,按照9∶1的比例分配训练数据(traindata)、验证数据(testdata).
根据结果标题对串联质谱数据进行肽序列匹配处理,通过质荷比进行强度匹配,获取肽序列离子碎片及离子强度信息.根据文献[9]介绍,碎片离子中b与y所占的强度比较大,文献[12]中介绍了选取b/y离子的可靠性,因此为了方便计算,这里只选取b型与y型离子,并根据肽序列算出每个碎片离子的质量,这里设定质量的误差范围为±0.02,在此范围内的离子选取离子强度最大的一个保存.
理论CID断裂,模拟肽序列FLGK如图1所示.
简化肽序列的断裂模型,假定只产生1电荷离子,b1∶F,b2∶FL,b3∶FLG,y1∶k,y2∶GK,y3∶LGK.计算离子质荷比公式(4)如下:
b离子=(氨基酸残基分子量+H*z)/z
y离子=(氨基酸残基分子量+H2O+H*z)/z
母离子=(所有氨基酸分子量+H2O+H)/z
(4)
表1pFind鉴定结果

数据集鉴定结果覆盖率/%FDR=0.1%/个去重/个母离子电荷为2时/%不含氧化修饰的数据/%处理后数据/个Hela数据集61.25873164279467.5480.2126604Yeast数据集57.41861543081368.5572.8419440
表2FDTK碎裂离子质荷比

离子残基质荷比离子残基质荷比b1F148.0762337-b2FD263.1031722y3DTK363.1879568b3FDT364.1508458y2TK248.1610183-y1K147.1133447
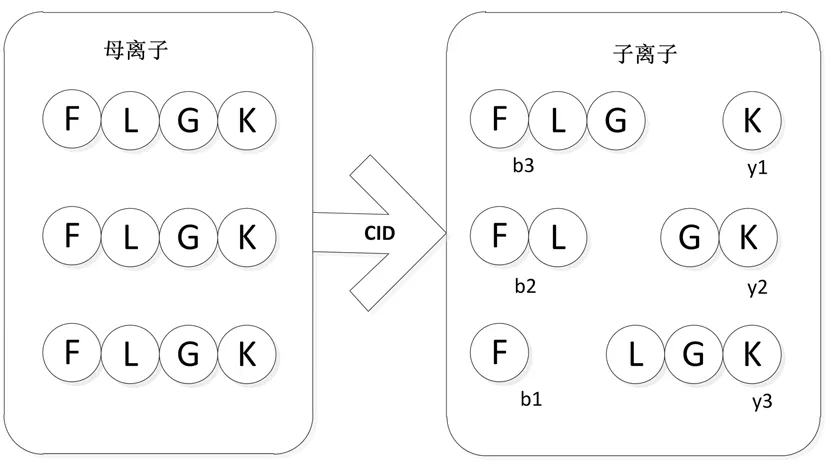
图1 模拟肽序列(FLGK)CID断裂
其中z为离子电荷量,本实验中选取的b与y离子都为1电荷,母离子电荷数为2.
表2以多肽序列FDTK为例,使用质荷比公式描述1电荷b离子与1电荷y离子的质荷比计算.氨基酸残基分子量F: 147.0684087,D:115.0269385,T: 101.0476736,K: 128.0949557.H分子质量1.007825,H2O分子质量18.010564.
读取处理后数据列表,将获取的离子信息保存.接下来循环读取离子信息,根据之前定义的离子特征,获取对应离子特征值,将离子信息保存为libsvm格式文件.文献[13]中介绍离子的三个主要特征:最高峰位置;相邻氨基酸功能;肽组成的总体特征.其中使用的主要离子特征来源于文献[3]与文献[5],离子特征的计算以及实验使用的所有离子特征可在附件中查找.
2.1.3 离子强度建模
使用处理完成的libsvm格式文件进行模型的构建,使用XGBoost工具库进行建模.程序执行完成之后模型将会保存到xgb.model,而dump.raw.txt中将model文件保存为人工可识别的决策树.
XGBoost中各项参数设置为:树的最大深度为11,迭代计算次数:14,收缩步长:0.04,目标函数为:linear.
2.2 预测序列离子强度相似度结果
循环读取待预测的肽序列,使用预测模型预测离子强度,并对预测的离子强度与实验离子强度求相似度得分.实验中Hela使用2660条肽序列数据进行预测,Yeast使用1944条肽序列数据进行预测,使用公式(3)求出每一个序列的相似度得分,最终求出平均值.根据文献[13]中对肽序列分为3类:移动肽,非移动肽,部分移动肽.对实验结果分类统计,见表3、表4、表5.
表3 实验整体离子强度相似度
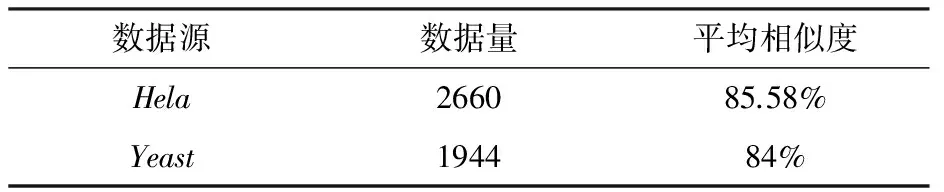
数据源数据量平均相似度Hela266085.58%Yeast194484%
由表3结果显示,使用GBDT算法构建的预测模型,其预测肽序列的离子强度与实验离子强度有着较高的相似度,可以提取碎片离子属性.
表4Hela数据碎裂离子强度相似度
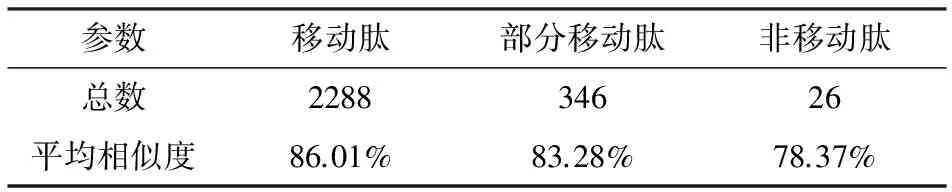
参数移动肽部分移动肽非移动肽总数228834626平均相似度86.01%83.28%78.37%
表5Yeast数据碎裂离子强度相似度

参数移动肽部分移动肽非移动肽总数16452936平均相似度84.39%82.14%70.71%
由表4、表5可以看出,肽序列断裂类型主要为移动肽,其次为部分移动肽,最后为非移动肽.前两种平均相似度较非移动肽有着更高的准确性且数量更多.根据Mobilepeptides的概念:肽序列中碱性氨基酸的数量小于母离子电荷数,因此可以初步估计,碱性氨基酸与母离子电荷数,是影响离子强度的主要特征之一.
并且分析预测产生的决策树,其中主要使用到的特征为:肽序列的质量、电荷,肽序列与碎片离子的疏水性、气相碱性、PI值、α螺旋的倾向,断裂位置,肽序列长度,碎片离子中组氨酸H、赖氨酸K、精氨酸R的比例等都对离子强度有着较大的影响,可以在肽谱匹配算法中给予这些特征以较高的权重.
本实验选取XGBoost作为模型的构建工具,其优点是预测速度快,并可以自动对离子特征的优先级进行排序,结果的整体相似度基本达到了85%,但仍有一定的提升空间.分析原因在于pFind鉴定结果的覆盖率未达到70%以上,并且在鉴定时打分算法只是取出最有可能的匹配,因此结果存在一定误差;另外模型构建时离子强度的选择数量不够,由于简化了实验碎片离子的选取,因此部分离子特征没有选择到,在后续的实验中,会加入其它的碎裂离子与离子特征进行建模,提高相似度.
3 结束语
本文使用梯度提升算法工具XGBoost构建肽序列的离子强度模型,并对实验步骤进行了详细的描述.最后针对具体肽序列使用文献中查找到的相似度指标来进行评估.对提取的特征可以应用到蛋白质鉴定软件的肽谱匹配算法中,进一步提高肽识别的性能.本实验对CID裂解的肽段离子进行了简化的处理,对子离子只选择1电荷的b与y离子的数据,母离子选择2电荷的数据,在后续的试验中将会考虑多电荷的复杂情况.实验使用到的特征及氨基酸属性值可在http://pan.baidu.com/s/1pKTyiW7获取.
[1] 付岩, 贺思敏, 孙瑞祥,等. 串联质谱蛋白质鉴定的关键计算问题[J]. 信息技术快报, 2010,8(01):16-32.
[2]ZHANGZ.Predictionoflow-energycollision-induceddissociationspectraofpeptides[J].Analyticalchemistry, 2004, 76(14): 3908-3922.
[3]ELIASJE,GIBBONSFD,KingOD,etal.Intensity-basedproteinidentificationbymachinelearningfromalibraryoftandemmassspectra[J].Naturebiotechnology, 2004, 22(2): 214-219.
[4]TANGH.Amachinelearingapproachtopredictingpeptidefragmntationspectra[J].PacificSymposiumonBiocomputingPacificSymposiumonBiocomputing, 2006, 11:219-30.
[5]ZHOUC,BOWKERLD,FENGJ.Amachinelearningapproachtoexplorethespectraintensitypatternofpeptidesusingtandemmassspectrometrydata[J].BmcBioinformatics, 2008, 9(2):1-17.
[6]SUNS,YANGF,YANGQ,etal.MS-Simulator:PredictingY-IonIntensitiesforPeptideswithTwoChargesBasedontheIntensityRatioofNeighboringIons.[J].JProteomeRes, 2012, 11:4509-4516.
[7]CHENT,GUESTRINC.Xgboost:Ascalabletreeboostingsystem[J].arXivpreprintarXiv:1603.02754, 2016.
[8]SONGR,CHENS,DENGB,etal.eXtremeGradientBoostingforIdentifyingIndividualUsersAcrossDifferentDigitalDevices[C]//InternationalConferenceonWeb-AgeInformationManagement.SpringerInternationalPublishing, 2016: 43-54.
[9] 于长永, 王国仁, 毛克明,等. 一种新颖的蛋白质序列与其串联质谱的匹配打分算法[J]. 小型微型计算机系统, 2010, 31(03):404-407.
[10]朱思敏, 李华梅. 基于泊松分布模型的蛋白质串联质谱鉴定算法研究[J]. 云南民族大学学报(自然科学版),2016, 25(2):179-184.
[11]NEUHAUSERN,MICHALSKIA,COXJ,etal.Expertsystemforcomputer-assistedannotationofMS/MSspectra[J].Molecular&CellularProteomics, 2012, 11(11): 1500-1509.
[12] 王中胜. 基于支持向量机分类的b/y离子峰选取算法及肽序列标签生成算法的研究[D]. 北京:中国人民解放军军事医学科学院, 2007.
[13]FRANKAM.Predictingintensityranksofpeptidefragmentions[J].JProteomeRes, 2009, 8:2226-2240.
(编辑:姚佳良)
Peptidefragmentionintensitymodelingbasedongradientboostingdecisiontree
HUAIHao,LIUXue,ZHANGLong-bo,WANGXiao-dan
(SchoolofComputerScienceandTechnology,ShandongUniversityofTechnology,Zibo255049,China)
Inordertofindthepeptidefragmentionsattributesthathaveagreaterinfluenceontheproteinidentificationalgorithm,andimprovetheaccuracyofidentificationresults,theforecastmodelbasedonfragmentionsioncharacteristicandintensityinformationisbuilt.Firstly,weuseofpFindtoidentifytandemmassspectrum,thenfilterouttheneededpeptidesequences;secondly,wecalculatetheresultofm/zandtheattributesvaluesofions,andgettheintensitiesbymatchingm/z,thenusetheinformationofintensityandtheattributesvalueofionstobuildalibsvmformatfile,andthenbuildapredictionmodelthroughtheXGBoostwhichusingtheGBDTalgorithm;finally,weusethebuiltpredictionmodeltopredictthetheoryintensitiesofpeptidesequencesionswhichproducedbyprotein.Theexperimentalresultsshowthattheintensitieswegotfromthepredictionmodelhadahighersimilaritytotheexperimentalintensities.Meanwhile,theanalysisofthepredictionmodelcanfindtheruleofpeptidefragmentationfromthepredictiontree.andextractthepeptidefragmentionsattributesthathadlargerinfluenceontheintensities.
tandemmassspectrometry;peptidefragmentionintensity;gradientboostingdecisiontree;modeling
2016-06-23
怀浩,男,haoyuexihuai@126.com; 通信作者:张龙波,男,zhanglb@sdut.edu.cn
1672-6197(2017)02-0064-05
TP
A
