茶树分子标记辅助育种研究进展
2017-03-09王让剑孔祥瑞陈常颂余文权
王让剑,杨 军,孔祥瑞,陈常颂*,余文权
(1.福建省农业科学院茶叶研究所/国家茶树改良中心福建分中心,福建 福安 355015;2.福建省农业科学院,福建 福州 350000)
茶树世代周期长,是多年生异花授粉木本作物,多为二倍体,基因组大小约为3.02G且高度杂合[1]。目前茶树新品种选育的主要方法仍然是常规育种法,包括选择育种和有性杂交,该方法工作量大、效率低、时间长,已难以适应快速发展的市场对多样化茶树品种的客观需求。分子标记辅助选择(Marker Assisted Selection,MAS)可显著提高育种效率,已成为作物遗传育种领域的研究重点。发掘与茶树优良经济性状紧密连锁的分子标记是开展茶树分子标记辅助选择育种的基础,目前主要依靠QTL作图(Quantitative Trait Loci Mapping)与关联作图(Association Mapping)两种方法。近年来茶树QTL作图与关联作图相关研究正有条不紊的开展,本文拟简述QTL作图和关联作图在茶树分子标记辅助选择研究中的进展及存在的问题,并对其发展趋势进行展望。
1 茶树QTL作图研究进展
1.1 茶树遗传图谱构建
构建遗传图谱的关键步骤是建立合适的遗传作图群体及选择合适的图谱构建方法。茶树世代周期长、人工杂交结实率低,难以得到像水稻等一年生作物用于构建遗传图谱的BC群体(回交群体)、NIL群体(近等基因系群体);茶树自交不亲和,也很难得到F2群体、RIL群体(重组自交系群体),因而基于上述群体的作图理论均无法直接应用于茶树遗传图谱构建。茶树遗传组成高度杂合,许多遗传位点在F1代即发生分离,因此一般利用F1群体基于“拟测交”(Pseudo Testcross)方法构建遗传图谱[2],该方法在林木等多年生作物遗传图谱构建中应用较多[3-4]。
茶树遗传图谱研究起步相对较晚,但随着“拟测交”方法的应用以及DNA分子标记技术的发展,茶树分子遗传图谱的构建工作得以逐步进行。从已发表的茶树遗传图谱来看,其是伴随茶树分子标记发展的进程逐渐展开的,具有较明显的代际界线(见表1)。起初,茶树分子标记的开发是从显性标记开始的,因此早期茶树遗传图谱构建以RAPD等显性标记为主,虽然这类标记无需知晓基因组的序列信息,操作简便,可迅速作图,但这类标记是对基因组的随机扩增,稳定性较差,很难实际应用,且其扩增片段多位于基因组的非编码区,不能直接用于分离目的基因。随后,由于SSR标记具有多态性高、稳定性强、共显性的特点,在茶树遗传研究中备受青睐,伴随茶树SSR标记开发的逐渐深入,茶树遗传图谱的构建逐渐以SSR标记为主,但SSR标记需要对其多态性进行验证,开发成本较高,费时费力。近年来,随着功能基因组研究的逐步开展,茶树基因组与转录组大规模测序成为现实,获得大量的SNP标记已比较容易。SNP标记具有高分型效率、数量巨大、全基因组覆盖、分析简单、成本较低等特点,可以预测SNP标记将会成为今后茶树遗传图谱构建的主要分子标记。
1.2 茶树QTL定位
进行QTL定位的遗传图谱,其标记的平均图距宜在10 cM以下,倘开展基因图位克隆研究,则目标区域的平均图距应在1 cM 以下[21-24]。早期构建的茶树遗传图谱其密度基本达不到要求,且均匀性不好,因此难以开展QTL定位工作。近年来,随着高通量测序技术的进一步发展及测序成本的持续降低,利用SSR、SNP等共显性标记构建符合QTL定位要求的茶树遗传连锁图谱已陆续报道出来,有关茶叶芽叶性状、儿茶素含量、抗病(虫)性状等QTL定位工作正稳步推进,并且取得了一些阶段性的成果,这为开展茶树分子标记辅助选择育种奠定了基础。
Tan等[15]利用茶树花转录组开发了2439个SSR标记并验证了其中的720个,成功开发出431个SSR标记,利用其中的237个标记对龙井43(♀)×白毫早(♂)的F1群体构建了一张茶树遗传图谱,包括15个连锁群,总图距为1156.9 cM,标记间平均距离为5.2 cM。随后,该课题组继续整合483个SSR标记,利用上述群体重新构建了一张图谱,包括15个连锁群,总图距为1226.2 cM,标记间平均距离为2.5 cM,且定位了15个与春茶萌芽期,嫩梢颜色,成熟叶长,成熟叶宽以及叶形指数相关的QTL[19]。Ma等[16]使用406个SSR标记,运用“拟测交”策略对迎霜(♀)×北跃(♂)单株的F1群体构建了一张茶树遗传图谱,包含15个连锁群,总图距为1143.5 cM,标记间平均距离为2.9 cM,定位了25个与儿茶素含量相关的QTL,其中9个QTL年份之间表现稳定,分布在LG03、LG11、LG12、LG15等连锁群上,每个QTL对群体变异的解释率在2.4%~71.0%之间。随后,该课题组又利用SLAF-seq简化基因组测序技术开发了茶树SNP标记,并利用6042个SNP标记对前期构建的图谱进行了加密,最终构建的图谱包括6448个标记,总图距为3965 cM,标记之间平均遗传距离为1.0 cM,该图谱是第一个基于SNP标记的茶树遗传图谱,也是目前最饱和的一张图谱[18]。徐礼羿[20]等以乌牛早(♀)×龙井43(♂)的F1群体,构建了一张双亲整合的遗传图谱,该图谱拥有16个连锁群,包含175个SSR标记,图谱总长为1165.4 cM,平均图距6.7 cM。在LG03、LG06、LGll、LGl3连锁群上共检测到4个的茶橙瘿螨抗性QTL,单个QTL的表型变异贡献率的变幅为7.2%~13.0%,且LG11存在1个控制茶橙瘿螨抗性的主效QTL,变异贡献率为13.0%。在LG01、LG04、LG06共检测到4个的日灼病抗性QTL,单个QTL的表型变异贡献率的变幅为6.9%~69.8%,且LG01和LG04各存在1个茶日灼病抗性相关的主效QTL,变异贡献率分别为16.5%、69.8%。在LG03、LG06、LG08、LG10、LG13、LG15连锁群上共检测到8个的炭疽病抗性QTL,单个QTL的表型变异贡献率的变幅为5.6%~13.8%,且LG10存在1个茶炭疽病抗性相关的主效QTL,变异贡献率分别为13.8%。
2 茶树关联作图研究进展
关联作图是以自然群体为研究对象,以长期重组后保留下来的标记基因间连锁不平衡(Linkage Disequilibrium,LD)为依据,将目标性状表型的多样性与标记基因型的多态性结合分析,可直接鉴定出与表型变异密切相关的标记基因位点[25-26]。相比QTL作图,关联作图不用构建人工杂交的作图群体,而且不存在QTL作图时两亲本基因型的限制,可同时检测同一座位的多个等位变异,并且定位更精确,甚至可以达到单基因的水准[27]。关联作图主要包括全基因组关联作图和候选基因关联作图两种方法。

表1 已构建的茶树遗传图谱
注:“-”表示没有。
2.1 全基因组关联作图
当选取的标记数目可以覆盖全基因组时,即可定位到所有影响表型的QTL,这种方法称为全基因组关联作图,该方法所需标记的数目取决于物种的基因组大小以及LD水平,物种基因组大小相近时,LD衰减速度慢的物种所需标记少,其定位精度比衰减速度快的物种低[28]。
随着SSR等标记的快速发展,茶树全基因组关联作图研究开始了一些积极的尝试。姚明哲等[29]利用自主开发的55个EST-SSR标记对112份茶树品种(系)进行了基因分型,通过关联分析共鉴定出5个与表型性状显著关联的EST-SSR标记(P<0.01),其中标记CS298、CS317、CS84分别与一芽二叶百芽重(BW)、叶片长度(LL)和茶多酚含量(TP)显著关联,对表型变异的解释率分别为0.11、0.15和0.35;标记CS330和CS338均与咖啡碱含量(CAF)显著关联,对表型变异的解释率分别为0.14和0.15。乔婷婷等[30]在110 个EST-SSR标记中共检测到4656个成对位点组合存在连锁不平衡(LD),占所有可能成对位点组合的38.8%,其中301个成对位点组合存在较高程度的连锁不平衡,39个标记与累计15个性状表现为极显著。苏会[31]利用EST-SSR标记对豫南茶区115个茶树种质进行了标记基因型分型,并将标记与茶品质相关性状进行了关联分析,结果表明,67对EST-SSR组成的共2211对成对位点组合中,259个成对位点连锁不平衡程度较高,占所有位点的11.71%。当K=4时,模型后验概率LnP(D)最大,群体被分为四个亚群。与水浸出物关联的标记有5个,其中TM066解释率最高,为13.32%;与茶多酚关联的标记有5个,其中TM092解释率最高,为13.68%;与氨基酸关联的标记有6个,其中TM083解释率最高,为7.6%;与咖啡碱关联的标记有3个,其中TM111解释率最高,为7.8%;TM066、TM092、TM074-2与水浸出物、茶多酚同时关联;TM086-1与茶多酚、氨基酸同时关联;TM088、TM111、TM124与氨基酸,咖啡碱同时关联。
2.2 候选基因关联作图
当主效基因或者质量性状基因对表型有决定性的影响,甚至单个碱基的变异亦可决定表型时,对可能影响表型性状的部分基因组区段进行关联分析,不用过多的基因型分析工作即可定位目的基因,这种方法称候选基因关联作图。应用全基因组关联作图方法研究LD衰减速度快的作物时,标记与QTL处于LD状态的概率相对较低,定位到目标基因的概率相对较小,这时采用候选基因关联作图法对这类作物进行分析效果更好[32]。利用候选基因关联作图法可鉴定到影响表型的基因组区段的多态性,并可估计其效应值,因此该方法可对特定基因的等位变异是否控制目标性状进行进一步验证,从而发掘出优异的等位基因[33-36]。候选基因关联作图法由于具有实验的目的性更明确,基因型分析成本更低等优点,因此备受研究者青睐。候选基因的选择主要包括生理生化途径中重要的功能基因、QTL研究定位区域所含的基因以及近缘物种研究中效应较大的同源基因等[37-42]。
茶树候选基因关联作图研究亦开始了一些积极的探索。Jin等[43]利用候选基因关联作图法研究了咖啡碱合成酶Ⅰ与咖啡碱含量之间的遗传关系,通过对咖啡碱合成酶Ⅰ基因重测序从44个茶树种质资源中共鉴定出87个SNP,从序列变异中鉴定出2个CAPS标记,其中SNP4318在4个环境中与咖啡碱含量相关,表型变异结实率为4.0%~7.7%。随后,该课题组又运用混合分离转录组测序(BSA-Seq)及候选基因关联作图法,在茶树中鉴定出了一个影响儿茶素合成的重要功能基因类黄酮3’,5’-羟化酶基因(F3’5’H),该基因位点有10个SNP标记与儿茶素指数CI、儿茶素组分含量、儿茶素总量等性状显著相关,并且基于F3’5’H中的SNP848标记开发了一个能鉴定和筛选高二羟基儿茶素茶树资源的功能标记[44]。该项研究为今后茶树重要性状的遗传解析及功能性分子标记开发提供了新的思路(如图1所示)。
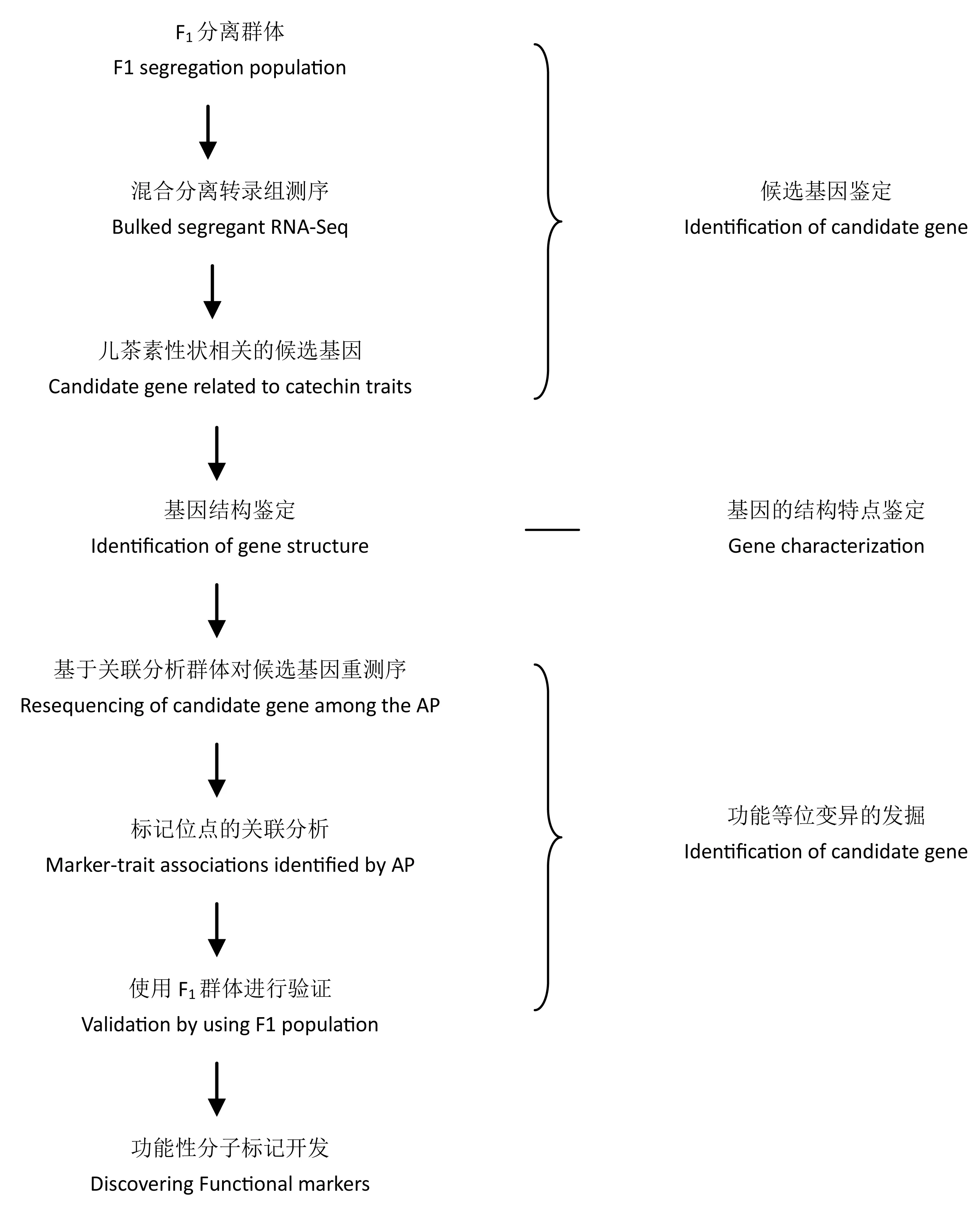
图1 茶树重要性状遗传解析流程图Fig.1 Strategic procedure for analyzing critical genetic traits of tea plants
3 存在的问题与展望
3.1 茶树QTL作图
当前多种作物在QTL作图方面已经取得了重要进展[45-47],并成功地应用于主效基因的导入和聚合,育成的水稻、玉米、大豆等大田作物品种亦在生产上得到了应用[48-50],相较而言,茶树QTL作图研究尚处起步阶段,利用该方法开展标记辅助选择育种研究面临着巨大的挑战。具体表现在:(1)茶树人工杂交周期长,从授粉到果实成熟需要1年时间,结实率非常低,目前用于构建茶树遗传图谱所用的群体均不超过200个个体,难以满足构建高密度遗传图谱对群体数目的要求。研究表明,当作图群体大小一定,仅靠增加标记数量并不能提高QTL作图的效率,只有群体个数达到400个以上时,检测到的QTL效应才趋于稳定[51]。(2)已构建的多数茶树遗传图谱选择材料构建群体时,考虑较多的是图谱绘制,而在实际育种应用上考虑得较少,且选用的作图群体不尽相同、标记类型多样,很难将多个图谱整合加以利用,此外,多数遗传图谱使用的标记为RAPD、AFLP等显性标记,重现性和保守性较差,远远达不到标记辅助选择育种的要求。(3)茶树遗传基础研究相对薄弱,例如:茶树性状的遗传力、性状之间的上位性效应以及QTL与环境互作等方面的研究基本处于空白阶段,这就会导致一些QTL效应估计不准确或QTL检测不到,进而对QTL定位的准确性产生较大的影响。
3.2 茶树关联作图
关联作图法虽然具有不用构建人工杂交的作图群体,可同时对多个等位基因进行分析,定位QTL精度更高等优点,但关联作图法亦存在一些缺陷。具体表现在:(1)关联作图群体中的LD受到遗传漂变、群体结构以及自然选择等诸多因素的影响,因此一些无关等位基因亦可与QTL形成LD,从而表现出与性状的伪关联或假阳性。研究表明,群体结构可解释表型性状中约9.3%的变异[52]。(2)关联作图虽对QTL的检测能力较高,但不能估计QTL在染色体上的具体位置及其加性、上位性效应,因此关联作图结合QTL作图结果一起分析效果才更好。
3.3 展望
利用QTL作图及关联作图开展茶树分子标记辅助育种研究,今后应加强以下工作:(1)在亲本选配时,尽量选择与育种直接相关的材料,构建的群体尽量做到既是遗传研究群体,也是育种群体,缩短QTL定位研究与育种实际应用的距离。(2)根据育种目标,选择合适的QTL作图群体及分子标记类型,构建高密度的茶树饱和遗传图谱,开展QTL精细定位与QTL克隆研究。(3)广泛收集、保存、鉴定各类茶树种质资源,开展茶树基因组、转录组、代谢组以及生物信息学研究,加强茶树次生代谢物质代谢网络及其重要功能基因研究。(4)相关单位协作,加强茶树性状的遗传力,QTL与QTL间、QTL与环境间的互作研究等。
[1]En-Hua xia, Hai-Bin Zhang, Jun Sheng,etal. The Tea Tree Genome Provides Insights into Tea Flavor and Independent Evolution of Caffeine Biosynthesis[J]. Molecular Plant,2017,10(6):866-877.
[2]马建强,姚明哲,陈亮.茶树遗传图谱研究进展[J].茶叶科学,2010,30(4):329-335.
[3]Brondani R P, Williams E R, Brondani C,etal. A microsatellite-based consensus linkage map for species of Eucalyptus and a novel set of 230 microsatellite markers for the genus[J]. BMC Plant Biol,2006,6(1):20.
[4]Doucleff M, Jin Y, Gao F,etal. A genetic linkage map of grape, utilizing Vitis rupestris and Vitis arizonica[J]. Theor Appl Genet,2004,109(6):1178-1187.
[5]田中淳一.RAPD をべースにしたチャの连锁地图の作成と遗传解析への利用の可能性[J].茶業研究報告,1996,84(别册):44-45.
[6]Ota S, Tanaka J. RAPD-based linkage mapping using F1segregating populations derived from crossings between tea cultivar ‘Sayamakaori’ and strain ‘Kana-Ck17’[J]. Breeding Research,1999,1(1):16.
[7]Hackett C A, Wachira F N, Paul S,etal. Construction of a genetic linkage map forCamelliasinensis(tea) [J]. Heredity,2000,85(4):346-355.
[8]黄建安,李家贤,黄意欢,等.茶树AFLP分子连锁图谱的构建[J].茶叶科学,2005,25(1):7-15.
[9]黄福平,梁月荣,陆建良,等.应用RAPD 和ISSR 分子标记构建茶树回交1代部分遗传图谱[J].茶叶科学,2006,26(3): 71-176.
[10]Mewan K M, Saha M C, Konstantin C,etal. Construction of a genomic and EST-SSR based saturated genetic linkage map of tea (CamelliasinensisL.) [C] // Proceedings of the 3rdInternational Conference on O-Cha (tea) Culture and Science (ICOS), Shizuoka, Japan:2007.
[11]Taniguchi F, Tanaka J, Kono I,etal. Construction of genetic linkage map of tea using SSR markers[C]//Proceedings of the 3rdInternational Conference on O-Cha (tea) Culture and Science(ICOS), Shizuoka, Japan: 2007.
[12]Kamunya S M, Wachira F N, Pathak R S,etal. Genomic mapping and testing for quantitative trait loci in tea (Camelliasinensis(L.) O. Kuntze) [J]. Tree Genetics & Genomes,2010,6(6):915-929.
[13]Chih-Yi Hu, Tair-Chyang Lee, Hsien-Tsung Tsai,etal. Construction of an integrated genetic map based on maternal and paternal lineages of tea(Camelliasinensis)[J]. Euphytica,2013,191(1):141-152.
[14]Taniguchi F, Furukawa K, Ota-Metoku S,etal. Construction of a high-density reference linkage map of tea (Camelliasinensis) [J]. Breeding Science,2012,62(3):263-273.
[15]Li-Qiang Tan, Li-Yuan Wang, Kang Wei,etal. Floral Transcriptome Sequencing for SSR marker Development and Linkage Map Construction in the Tea Plant (Camelliasinensis) [J].PLOS ONE, 2013,8(11): e81611.
[16]Jian-Qiang Ma, Ming-Zhe Yao, Chun-Lei Ma,etal. Construction of a SSR-Based Genetic Map and Identification of QTLs for Catechins Content in Tea Plant(Camelliasinensis) [J]. PLOS ONE, 2014,9(3): e93131.
[17]Sapinder Bali, Akshay Mamgain, Soom Nath Raina,etal. Construction of a genetic linkage map and mapping of drought tolerance trait in Indian beveragial tea[J]. Mol Breeding,2015(35):112.
[18]Jian-Qiang Ma, Long Huang, Chun-Lei Ma,etal. Large-Scale SNP Discovery and Genotyping for Constructing a High-Density Genetic Map of Tea Plant Using Specific-Locus Amplified Fragment Sequencing (SLAF-seq)[J]. PLoS ONE,2015,10(6):e0128798.
[19]Li-Qiang Tan, Li-YuanWang, Li-Yi Xu,etal. SSR-based genetic mapping and QTL analysis for timing of spring bud flush, young shoot color, and mature leaf size in tea plant (Camelliasinensis) [J]. Tree Genetics & Genomes,2016,12(5):52.
[20]徐礼羿.茶树SSR遗传连锁图谱构建及茶橙瘿螨、日灼病和炭疽病抗性QTL的定位[D].成都:四川农业大学,2016.
[21]Yu-Juan Zhong, Yang-Yang Zhou, Jun-Xing Li,etal. A high-density linkage map and QTL mapping of fruit-related traits in pumpkin (CucurbitamoschataDuch.) [J]. Scientific reports, 2017,7(1):12785.
[22]Fa Cui, Na Zhang, Xiao-li Fan,etal. Utilization of a Wheat660K SNP array-derived high-density genetic map for high-resolution mapping of a major QTL for kernel number [J]. Scientific reports.2017, (1):3788.
[23]Changyou Liu, Baojie Fan, Zhimin Cao,etal. Development of a high-density genetic linkage map and identification of flowering time QTLs in adzuki bean (Vignaangularis) [J]. Scientific reports.2016(6):39523.
[24]莫惠栋,顾世梁.基因组长度的估计方法[J].科学通报,2003,45(13):1414-1418.
[25]Gaut B S, Long A D. The lowdown on linkage disequilibrium[J]. Plant Cell,2003,15(7):1502-1506.
[26]Doerge R W. Mapping and analysis of quantitative trait loci in experimental populations[J]. Nat Rev Genet,2002,3(1):43-52.
[27]谭贤杰,吴子恺,程伟东,等.关联分析及其在植物遗传学研究中的应用[J].植物学报,2011,46(1):108-118.
[28]Flint-Garcia S A, Thornsberry J M, Buckler E S. Structure of linkage disequilibrium in plants[J]. Annu RevPlant Biol,2003,54(4):357-374.
[29]姚明哲,乔婷婷,马春雷,等.EST-SSR标记与茶树表型性状关联的初步分析[J].茶叶科学,2010,30(1):45-51.
[30]乔婷婷.茶树资源遗传多样性及其表型性状关联EST-SSR位点的初步鉴定[D]北京:中国农业科学院, 2010.
[31]苏会.豫南茶区茶品质相关性状与EST-SSR标记的关联分析[D].郑州:河南农业大学,2016.
[32]Eric R Gamazon, Heather E Wheeler, Kaanan P Shah,etal. A gene-based association method for mapping traits using reference transcriptome data[J]. Nature Genetics,2015,47(9):1091-1098.
[33]Caiping Cai, Shuang Wu, Erli Niu,etal. Identification of genes related to salt stress tolerance using intronlength polymorphic markers, association mapping and virusinduced gene silencing in cotton[J]. Scientific reports.2017,7(1):528.
[34]Liu Y, He Z, Appels R,etal. Functional markers in wheat: current status and future prospects[J]. Theor Appl Genet,2012,125(1):1-10.
[35]Wangzhen Guo, Caiping Cai, Changbiao Wang,etal. A microsatellite-based, gene-rich linkage map reveals genome structure, function and evolution in Gossypium[J]. Genetics.2007,176(1):527-541.
[36]X Li, W Gao, H Guo,etal. Development of EST-based SNP and InDel markers and their utilization in tetraploid cotton genetic mapping[J]. BMC Genomics,2014,15(1):1046.
[37]Wang C, Ulloa M, Mullens T R,etal. QTL analysis for transgressive resistance to root-knot nematode in interspecific cotton (Gossypiumspp.) progeny derived from susceptible parents[J]. PLoS One,2012,7(4):e34874.
[38]Alfred Q, Liu HY, Xu HM,etal. Mapping of quantitative trait loci for oil content in cottonseed kernel[J]. J Genet, 2012,91(3):289-295.
[39]J Yu, K Zhang, S Li,etal. Mapping quantitative trait loci for lint yield and fiber quality across environments in aGossypiumhirsutum×Gossypiumbarbadensebackcross inbred line population[J]. Theor Appl Genet,2013,126(1):275-287.
[40]DD Fang, JN Jenkins, DD Deng,etal. Quantitative trait loci analysis of fiber quality traits using a random-mated recombinant inbred population in Upland cotton (GossypiumhirsutumL.)[J]. BMC Genomics,2014,15(1):397.
[41]Cao Z, Wang P, Zhu X,etal. SSR marker-assisted improvement of fiber qualities in Gossypium hirsutum using G. barbadense introgression lines[J]. Theor Appl Genet,2014,127(3):587-594.
[42]Said JI, Lin Z, Zhang X,etal. A comprehensive meta QTL analysis for fiber quality, yield, yield related and morphological traits, drought tolerance, and disease resistance in tetraploid cotton[J]. BMC Genomics,2013,14(1):776.
[43]Ji-Qiang Jin, Ming-Zhe Yao, Chun-Lei Ma,etal. Association mapping of caffeine content with TCS1 in tea plant and its related species[J]. Plant Physiology and Biochemistry,2016,105:251-259.
[44]Ji-Qiang Jin, Jian-Qiang Ma, Ming-Zhe Yao,etal. Functional natural allelic variants of flavonoid 3’,5’-hydroxylase gene governing catechin traits in tea plant and its relatives[J]. Planta,2017,245(3):523-538.
[45]Teresa B, De Leon, Steven Linscombe,etal. Molecular Dissection of Seedling Salinity Tolerance in Rice (OryzasativaL.) Using a High-Density GBS-Based SNP Linkage Map[J]. Rice,2016,9(1):52.
[46]Changlin Liu, Qiang Zhou, Le Dong,etal. Genetic architecture of the maize kernel row number revealed by combining QTL mapping using a high-density genetic map and bulked segregant RNA sequencing[J]. BMC Genomics,2016,17(1):915.
[47]Zhaoming Qi, Junbo Pan, Xue Han,etal. Identification of major QTLs and epistatic interactions for seed protein concentration in soybean under multiple environments based on a high-density map[J]. Mol Breeding,2016,36(5):55.
[48]万建民.中国水稻分子育种现状与展望[J].中国农业科技导报,2007,9(2):1-9.
[49]李建生.玉米分子育种研究进展[J].中国农业科技导报,2007,9(2):10-13.
[50]邱丽娟,王昌陵,周国安,等.大豆分子育种研究进展[J].中国农业科学,2007,40(11):2418-2436.
[51]苏成付.大豆QTL准确定位技术和策略的研究[D].南京:南京农业大学,2009.
[52]Flint-Garcia SA, Thuillet A, Yu J,etal. Maize association population: a high resolution platform for quantitative trait locus dissection[J]. Plant J,2005,44(6):1054-1064.
