多步骤决策树方法在基因表达数据上的应用研究*
2017-03-09哈尔滨医科大学卫生统计学教研室150081马李冰李贞子
哈尔滨医科大学卫生统计学教研室(150081) 马李冰 侯 艳 李贞子 李 康
多步骤决策树方法在基因表达数据上的应用研究*
哈尔滨医科大学卫生统计学教研室(150081) 马李冰 侯 艳 李贞子 李 康△
基因芯片技术得到的基因表达谱数据具有维数高、噪声大、样本量小、非线性等特点,如何从高维数据中提取含在其中的生物学信息,是医学和生物学研究中面临的一个重大挑战。基因表达数据分析的重要任务是筛选差异表达基因及对基因或样品进行分类,通过比较正常和疾病状态下基因表达的差异,研究疾病的发病机制、早期诊断和治疗方法。
目前用于高维组学数据的变量筛选方法主要有单变量筛选和多变量筛选[1],常用的单变量筛选方法有Satterthwaitt检验、SAM(significance analysis of microarrays)法、Wilcoxon秩和检验等;多变量筛选方法有随机森林(random forest,RF)、Boost方法等方法。单变量和多变量筛选各有优缺点,单变量筛选方法简单而快速,独立于判别模型,但是不考虑变量之间的相关关系;多变量筛选方法则能够考虑变量之间的交互作用。然而,任何一种多变量分析方法都有其局限性,如其适用范围和分析重点不同,为此可以把不同的分析方法结合在一起,对数据进行分析,可以得到更为丰富和可靠的结果。
本文在ClarLynda[2]等提出的多步骤分析策略的基础上,给出了一种新的多步骤决策树分析方法,这种方法将不同的数据降维方法有机融合在一起,先对数据进行变量初筛,然后对筛出来的差异变量做指标聚类,对每一类做主成分分析,用几个主成分基因进行判别分析。多步骤决策树经过多次降维,使数据维数灾难大大降低,提高的数据分析的效能,决策树判别分析也为疾病分型提供依据,逆向寻找各主成分相关的差异基因,可对疾病的发病机制提供有效的信息。本文在简要介绍多步骤决策树方法的基础上,通过实际数据分析,探索其准确性,并与单变量筛选SAM和多变量筛选RF进行比较。
原理与方法
多步骤决策树(multistep decision tree,MDT)是针对高维组学数据的一种筛选方法,即将变量筛选、变量聚类和主成分分析结合在一起的分析方法,主要有四个连续的步骤组成,流程如图1所示,具体分析过程如下:

图1 多步骤决策树分析流程图
1.用SAM方法筛选差异基因
首先使用SAM对原始的基因表达矩阵进行单变量筛选,得到“差异基因”。SAM变量筛选的阈值选择q≤0.05,q≤0.05的基因即为有意义的差异基因,其中q为经FDR(false discovery rate,错误发现率)校正后的P值。
SAM是目前被认为较好的基因筛选方法[3],其基本思想就是在传统的t检验公式的分母上加上一个较小的正数S0(S0是能够使变异系数最小的值),使构建的统计量在分子(均数差值)较小的情况下不容易得到较大的值[4]。针对每个基因i,d(i)能够反映基因表达强度与类别之间的关系。具体的算法如下:
(1)

(2)
(3)

2.变量聚类分析
为了对数据进一步进行降维,对上述筛选得到的差异基因做变量聚类分析。变量聚类是依据变量之间的距离,把可能相关的变量聚为一类。通常有两种变量聚类方法:一种是用变量的相关矩阵进行聚类,另一种是用因子分析或者主成分分析得到的变量结构进行聚类。最常用的是第一种,即先计算变量之间的距离矩阵(如,相关矩阵),然后对相关系数矩阵做聚类,最后获得同质的聚类[5]。
本研究采用相关矩阵进行变量聚类。首先对差异基因矩阵做相关分析,然后计算相关矩阵的欧几里得(Euclid)距离,用最长距离法(completelinkagemethod)将相关的差异基因聚类,聚类的结果为6类,分别记为类clustA,clustB,……,clustG。
3.主成分分析
为了将不同聚类类别的基因作为整体进行判别分析,降低维数灾难,本研究进一步对每个聚类类别的基因做主成分分析,构建主成分基因。以碎石图为依据选择最佳主成分(PCs)个数,所有能够解释该类基因50%方差的主成分都会被选择,每个主成分基因(metagenes)是该聚类中的基因表达变量与其载荷的矩阵相乘。如聚类3中有2个主成分基因,分别记为clustC-1,clustC-2。
4.决策树
决策树是一种基于信息论的直观快速分类方法,将对象空间划分为若干子集。目前决策树方法中比较流行的算法有ID3、C4.5、CART和SPRINT等[7]。其中最具有代表性的是Quinlan提出的C4.5算法[8]。C4.5算法是ID3的改进算法,该算法根据信息增益率(informationgainratio)来选择变量,改善了ID3算法用信息增益选择属性的缺点,同时C4.5能对连续属性进行离散化处理,克服了ID3只能处理离散变量的不足。
信息增益率指信息增益与初始信息量的比值[7],对于样本集T,设样本量为n,共有m个类别,类别i在总样本集中出现的概率Pi,I(T)为样本集T的信息熵,那么样本集T的信息熵是:
(4)
假设根据变量A将样本集T划分为v个子集,其中子集Tj包含的样本个数为nj,则划分后的熵为
(5)

为了观察主成分基因对疾病的贡献大小,以及对数据分类判别的效果,可以在构建主成分基因的基础上研究疾病分型,并结合生物学功能数据库研究发病机制。本研究采用C4.5算法根据研究对象状态构建决策树,使用前剪枝法进行决策树修剪,修剪规则是每个终点叶上至少包含总的研究对象的10%,即最小实例数(minNumObj)不小于总样本数的10%。为避免过拟合,在此对层数不做限制,采用十折交叉验证(cross-validation)的方法进行判别分析,并计算灵敏度、特异度和信息比,评价判别模型的预测效果。
实例分析
为研究多步骤决策树方法在实际高维基因表达数据上的效果,选取3个卵巢癌基因表达数据进行分析,数据的基本情况如表1。为与单独使用一种的变量筛选方法比较,分别对多步骤决策树、SAM和随机森林筛选出来的变量构建决策树判别模型,比较其在基因表达数据上的分析效果和预测的准确性。

表1 三个卵巢癌基因表达数据的样本分布情况
1.GSE12470数据分析
多步骤决策树分析:首先用SAM进行变量筛选,筛选出健康、早期、晚期差异基因256个;对这256个差异基因进行指标聚类,聚为6类,分别记为ClusterA,…,ClusterF;然后分别对这6个类别进行主成分分析,按照贡献率大于50%,每一类最佳主成分个数分别是1,1,1,1,1,2,将各主成分载荷分别与原始变量矩阵相乘,产生出7个主成分基因,分别记为ClusterA-1,ClusterB-1,ClusterC-1,ClusterD-1,ClusterE-1,ClusterF-1,ClusterF-2。
SAM分析:单变量筛选用SAM方法,依然选用q≤0.05的变量,筛选出健康、早期、晚期差异基因256个。
RF分析:多变量筛选选择随机森林,随机森林树设置为500(ntree=500),每个分裂点样本预测个数设置为(mtry=148),分别选择前50,100,200,300个差异基因。
使用十折交叉验正方法对决策树模型进行评价。根据研究对象状态共构建了5棵决策树,分别分析这些主成分基因对健康、早期、晚期的判别效果,以及不同两类的分类效果,并选择灵敏度、特异度、信息比作为评价指标,结果如表2所示。
图2绘出了GSE12470数据分析判别的灵敏度和特异度。结果表明,在灵敏度、特异度上,多步骤决策树通过多步降维得到主成分基因的判别分析的效果多数情况下优于单纯的SAM分析和随机森林分析,信息比结果也显示,多数情况使用多步骤决策树方法建立的模型能提供更多的预测信息。
图3为GSE12470数据分析得到的决策树图。由图可知,在区分不同疾病状态时,不同的主成分基因对疾病状态的作用不同。聚类A的主成分基因能够区分健康对象和早期卵巢癌患者,聚类B能够区分健康对象和晚期卵巢癌患者,聚类E能够区分早期卵巢癌和晚期卵巢癌患者。
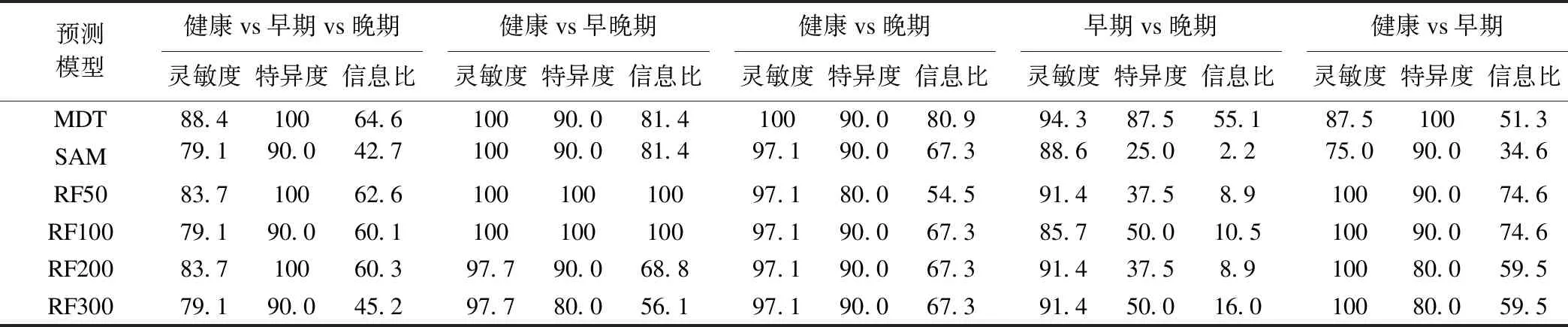
表2 GSE12470数据分析的判别效果比较

图2 GSE12470数据分析判别灵敏度、特异度、信息比

图3 GSE12470决策树图
为研究A、B和E这3个聚类所包含的差异基因,可以逆向寻找构成这些主成分基因的差异基因,如在KEGG中寻找这些差异基因的通路,并在文献中查找该基因是否与卵巢癌相关。表3结果显示,在能查到通路的差异基因中,聚类A有33.33%的基因在同一通路,聚类B中有45.45%的基因在同一通路,聚类E有38.46%的基因在同一通路,通过查阅文献,上述每类中都有确定与卵巢癌发病机制相关的基因,并有一些基因与其他的癌症相关,这些基因有可能是卵巢癌的潜在标志物。

表3 聚类A,B,E中的基因通路查询结果
2.GSE18520和GSE26712数据分析
为验证多步骤决策树分析数据的效果,同时分析了第二个和第三个基因表达数据。
对于数据GSE18520,SAM筛选出差异基因3206个,聚为6类,进行主成分分析后,对主成分基因进行决策树建模。随机森林筛选变量后,分别取前 500,
1000,2000,3000,3206,用决策树建模。对于数据GSE26712,SAM筛选出差异基因3539个,聚为6类,用相同的方法进行分析(RF取500,1000,2000,3000,3539个基因)。模型评价用十折交叉验证方法,并用灵敏度、特异度和信息比对判别效果进行比较,结果如表4和图4。结果显示,多步骤决策树在这两组数据中的分析效果都较单纯SAM和单纯随机森林变量筛选方法更优。

表4 GSE18520和GSE26712分析判别效果比较
讨 论
分析基因组学数据,多步骤决策树方法有几个优点:首先,通过不同分析阶段对数据降维,能更好地揭示基因组学数据结构;其次,在各个分析阶段最大程度保留了数据变量的信息,使结果解释变得更加容易;再有,分析的最后一步构建的决策树对数据结构和分布无任何假定,可以较好地进行分类;最后,可以通过建立的预测模型逆向寻找构成主成分基因的各差异基因,并可以根据主成分基因的构造,结合生物数据库进一步研究基因的功能。三个实际数据分析都表明本文给出的方法较单一分析方法更为有效。多步骤决策树方法主要的局限性是多步骤决策树使用了不同方法,前面分析方法的效果会影响后面分析的效果。尽管如此,多步骤决策树方法提供了一种新的分析思路,目的是提高数据挖掘和分析的效率。

图4 GSE18520和GSE26712判别分析效果
[1]Saeys Y,Inza I,Larranaga P.A review of feature selection techniques in bioinformatics.Bioinformatic,2007,23(19):2507-2517.
[2]Williams-DeVane CR,Reif DM,Hubal EC,et al.Decision tree-based method for integrating gene expression,demographic,and clinical data to determine disease endotypes.BMC Systems Biology,2013,7:119.
[3]Tusher V G,Tibshirani R,Chu G.Significance analysis of microarrays applied to the ionizing radiation response.Proc Natl Acad Sci USA,2001,98:5116-5121.
[4]赵发林,闫晓光,李康.几种差异基因分析方法及筛选效果比较.中国卫生统计,2008,25(4):354-356.
[5]Bandyopadhyay S,Mukhopadhyay A,Maulik U.An improved algorithm for clustering gene expression data.Bioinformatics,2007,23(21):2859-2865.
[6]Wold S.Principle Component Analysis.Chemometrics and Intelligent Laboratory Systems,1987,2:37-52.
[7]陈安,陈宁,周龙骧.数据挖掘技术及应用.北京:科学出版社,2006.
[8]李楠,段隆振,陈萌.决策树C 4.5算法在数据挖掘中的分析及应用.计算机与现代化,2008,12(4):160-163.
[9]Quinlan JR.Induction of Decision Tree.Machine Learning,1986,1:81-106.
[10]Kosuke Yoshihara,Atsushi Tajima,et al.Gene expression profiling of advanced-stage serous ovarian cancers distinguishes novel subclasses and implicates ZEB2 in tumor progression and prognosis.Cancer Sci,2009,10(8):1421-1428.
[11]Mok SC,Bonome T,Vathipadiekal V,et al.A Gene Signature Predictive for Outcome in Advanced Ovarian Cancer Identifies a Survival Factor:Microfibril-Associated Glycoprotein 2.Cancer Cell,2009,16(6):521-532.
[12]Bonome T,Levine DA,Shih J,et al.A Gene Signature Predicting for Survival in Suboptimally Debulked Patients with Ovarian Cancer.Cancer Res,2008,68(13):5478-5486.
(责任编辑:郭海强)
国家自然科学基金资助(81302511,81473072);黑龙江省博士后资助经费(LBH-Z14174)
△通信作者:李康,E-mail:likang@ems.hrbmu.edu.cn
