利用CRISPR/Cas9n double nick系统构建人DNAH2基因敲除的U2OS细胞株
2017-03-02常丽贤孙聪聪陈晓娟杨文钰张家源张英弛袁卫平竺晓凡
常丽贤,孙聪聪,陈晓娟,杨文钰,张家源,张英弛,袁卫平,竺晓凡
中国医学科学院 北京协和医学院 血液病医院血液学研究所,天津 300020
利用CRISPR/Cas9n double nick系统构建人DNAH2基因敲除的U2OS细胞株
常丽贤,孙聪聪,陈晓娟,杨文钰,张家源,张英弛,袁卫平,竺晓凡
中国医学科学院 北京协和医学院 血液病医院血液学研究所,天津 300020
常丽贤, 孙聪聪, 陈晓娟, 等. 利用CRISPR/Cas9n double nick系统构建人DNAH2基因敲除的U2OS细胞株. 生物工程学报, 2017, 33(2): 284–293.
Chang LX, Sun CC, Chen XJ, et al. Knocking out of humanDNAH2gene in U2OS cells by CRISPR/Cas9n double nick system. Chin J Biotech, 2017, 33(2): 284–293.
基于CRISPR/Cas9n double nick技术构建人DNAH2(Homo sapiens dynein, axonemal, heavy chain 2) 基因敲除的U2OS稳定细胞株,旨在研究DNAH2基因的生物学功能。首先设计并合成A、B两个sgRNA (Single guide RNA) 以及各自的互补链,退火连接形成DNAH2 sgRNA-A、B双链,再分别与带有BbsⅠ粘性末端的pX462线性载体相连,形成pX462-DNAH2-A、pX462-DNAH2-B重组真核表达质粒。质粒共转染至U2OS细胞后,加入嘌呤霉素,以有限稀释法获得阳性单克隆细胞株,再以蛋白印迹实验检测DNAH2蛋白的表达,最后通过PCR-基因测序技术分析突变特点。结果显示A、B sgRNA双链成功插入pX462载体,U2OS-DNAH2-KO单克隆细胞株中DNAH2蛋白不表达,DNAH2基因发生移码突变,从而证实利用CRISPR/Cas9n double nick系统成功构建人DNAH2基因敲除的U2OS稳定细胞株,为研究DNAH2基因提供有利工具。
DNAH2,CRISPR/Cas9n,基因敲除,稳定细胞株
DNAH2(Homo sapiens dynein,axonemal,heavy chain 2) 基因可以编码DNAH2蛋白,后者主要表达于气管和睾丸部位,可参与精子鞭毛的组成,是呼吸道纤毛主要的动力产生蛋白[1-3]。DNAH2基因突变的患者可能会出现呼吸道抵抗力下降、反复呼吸道感染、支气管扩张症等症状[2-4]。范可尼贫血 (Fanconi anemia,FA) 是一种主要由FA基因纯合或复合杂合突变引起的常染色体或X连锁隐性遗传性疾病[5-7]。目前已发现并克隆出多个可致范可尼贫血的突变基因(如FANCA、FANCB、FANCD1、FANCD2等)[5-6]。本实验室前期对明确诊断为范可尼贫血的5例患者及其父母进行了全外显子组测序,结果发现5例患者均有已知FA基因突变,并且含有2个或2个以上FA基因的杂合突变,同时还发现了其他多个新的突变基因,其中包括DNAH2基因[8]。然而,目前尚无DNAH2基因与范可尼贫血的相关性研究。CRISPR/Cas9 (Clustered regulatory interspaced short palindromic repeat/Cas9) 系统是一种在sgRNA的指导下,在基因组水平上对目的基因DNA序列进行改造的定点编辑技术,可实现对于目的基因的完全敲除。本实验旨在利用CRISPR/Cas9系统建立DNAH2基因靶向敲除细胞株,有助于探讨DNAH2基因在范可尼贫血中的作用机制。
1 材料与方法
1.1 材料
U2OS细胞为本实验室冻存;trans1-T1感受态细胞、Taq酶、SDS上样缓冲液、0.2%通透液购自天津科仪嘉欣科技有限公司;真核表达质粒pSpCas9n(BB)-2A-Puro (pX462) 购自Addgene;限制性内切酶BbsⅠ购自Thermo Fisher Scientific公司;质粒保护核酸外切酶(PlasmidSafe exonuclease) 购自epicentre公司;T4 DNA连接酶购自NEB公司;DNA快速凝胶回收试剂盒购自北京博大泰克生物基因技术有限公司;嘌呤霉素购自北京索莱宝科技有限公司;质粒快速小提试剂盒购自北京索来宝科技有限公司;sgRNA序列及基因测序工作由苏州金唯智生物科技有限公司完成。去内毒素质粒提取试剂盒购自Promega公司;NeoFect DNA转染试剂购自零客创智有限公司;BCA蛋白测定试剂盒购自Pierce公司;鼠源单克隆抗DNAH2抗体为本实验室制备;鼠源单克隆抗β-actin蛋白抗体购自Sigma公司;辣根过氧化物酶标记的抗鼠源IgG二抗购自KPL公司;BeyoECL Plus (超敏ECL化学发光试剂盒) 购于碧云天公司。
1.2 方法
1.2.1 sgRNA寡核苷酸双链序列设计
应用http://crispr.mit.edu/网站针对DNAH2基因 (NM_020877.2) 第1个外显子区的序列“5ʹ-ATGTCCAGCAAAGCTGAGAAGAAGCAG CGATTGAGTGGCCGAGGAAGCTCCCAGGC AAGCTGGTCAGGGCGGGCCACTCGGGCTG CTGTGGCCACACAGGAGCAGGGGAATGCC CCGGCTGTCAGTGAGCCAGAGCTGCAGGCT GAGCTCCCCAAGGAGGAGCCTG-3ʹ”进行A、B两个sgRNA的靶点选择及序列设计。首先通过在线网站获取特定靶点的A、B两对sgRNA原始片段,再对A原始片段进行如下修改:1) 去掉由网站设计的sgRNA原始片段3′端的“NGG”;5′端添加“CACC”,若“CACC”后非“G”,可加入“G”,以增加酶切效率,从而获得DNAH2-sgRNA-A序列;2) 去掉由网站设计的sgRNA片段3′端的“NGG”;再以此为基础设计出反向互补序列;5′端添加“AAAC”后即为DNAH2-sgRNA-A序列的互补链。然后,依此法对B原始片段进行修改。由此便分别获得带有BbsⅠ酶切位点的DNAH2-sgRNA-A、DNAH2-sgRNA-B两种寡核苷酸双链,具体序列见表1。
1.2.2 DNAH2-sgRNA双链和线性pX462载体的获取
由公司合成上述的DNAH2 sgRNA序列及其互补链;利用Thermo cycler使sgRNA序列与其互补链退火连接形成DNAH2-sgRNA-A、B两个双链结构,并稀释至0.5 μmol/L。反应体系:100 nmol/L DNAH2-sgRNA-A/B,100 nmol/L sgRNA-A/B的互补链,1 μL 10×T4 连接缓冲液,100 U T4 PNK,加超纯水至10 μL。退火程序:95 ℃ 5 min;94℃ l min,–l ℃/循环,22个循环;72 ℃ 30 min;71 ℃ 1 min,–1 ℃/循环,46个循环;4 ℃ 1 h。同时,以质粒快速小提试剂盒提取pX462质粒,再以BbsⅠ限制性内切酶,37 ℃、15 min进行酶切线性化。反应体系:1 μg pX462载体,1 μLBbsⅠ,1 μLFastAP,2 μL 10×Green Fast Digest Buffer,加超纯水稀释至20 μL。以DNA快速凝胶回收试剂盒回收线性pX462载体。

表1 DNAH2 sgRNA (A-B) 寡核苷酸双链序列Table 1 The double-stranded oligonucleotide sequences of DNAH2 sgRNA (A-B)
1.2.3 pX462-DNAH2-sgRNA的连接、转化及鉴定
取50 ng线性化pX462质粒,1 μL DNAH2-sgRNA双链 (0.5 μmol/L),1.5 μL 10×T4 DNA连接酶缓冲液,0.5 μL 牛血清白蛋白 (10 g/L),再加超纯水至11 μL体系,室温孵育60 min。在上述体系中加入1.5 μL 10×质粒保护缓冲液,1.5 μL ATP (10 mmol/L),1 μL质粒保护核酸外切酶,37 ℃孵育30 min,目的是防止非特异的重组产物。然后,分别将连接的pX462-DNAH2-sgRNA-A、B重组质粒转化入trans1-T1感受态细胞中,挑取单克隆进行摇菌扩增;再以质粒快速小提试剂盒提取两个重组质粒pX462-DNAH2-A和pX462-DNAH2-B,分别采用U6启动子序列 (ATACGATACAAGGCTGT TAGAGAGATA) 进行测序验证,同时利用BbsⅠ限制性内切酶进行酶切鉴定。
1.2.4 细胞培养与质粒共转染
在6孔板内以高糖-DMEM培养基 (含10%胎牛血清),5% CO2、37 ℃常规培养U2OS细胞。以去内毒素质粒提取试剂盒提取无内毒素质粒,包括pX462-DNAH2-A、pX462-DNAH2-B及空载体对照pX462。当U2OS细胞达60%−70%汇合时,更换新鲜培养基,1 h后根据产品说明书以NeoFect转染试剂对U2OS细胞转染空载体对照pX462,以制备U2OS-DNAH2-WT细胞株;或共转染pX462-DNAH2-A、pX462-DNAH2-B质粒,以制备U2OS-DNAH2-KO细胞株。
1.2.5 细胞稳定株的单克隆筛选
以6孔板常规培养未转染的U2OS细胞,分别以加入20、10、5、2.5、1.25和0.62 μg/mL嘌呤霉素的培养基培养48 h,确定将所有未转染的U2OS细胞杀死的最低浓度为2.5 μg/mL。然后在上述细胞转染后48 h,更换培养基为含有2.5 μg/mL嘌呤霉素的新鲜培养基,继续培养72 h后撤去嘌呤霉素继续培养。待细胞长满时,以胰酶消化后铺到15 cm皿中,平均每个显微镜低倍镜视野中有3−5个细胞即可。待细胞长出单克隆集落时,以高压灭菌过的细棉签将单克隆集落轻轻刮取,分别移至96孔板中培养,并进行编号。待96孔板长满,移至24孔板中继续培养。
1.2.6 蛋白印迹实验鉴定
分别将上述各孔细胞再接种至6 cm皿中扩大培养。对同一单克隆来源的细胞分为两部分,一部分进行蛋白印迹鉴定,另一部分继续培养,用于鉴定成功后的细胞冻存。待细胞融合至80%−90%,先以1×磷酸缓冲液 (PBS) 溶液洗涤细胞3次,再加入预冷的1×NP-40裂解液裂解细胞,超声破碎细胞后4 ℃、12 000 r/min 离心10 min,取上清获得全细胞裂解液;再以BCA蛋白测定试剂盒测定总蛋白浓度;裂解液中加入SDS上样缓冲液,99 ℃热变性5 min,进行6% SDS-PAGE电泳。电泳结束后,以湿转法将凝胶上蛋白转移至PVDF膜,用含5%脱脂奶粉的TBST溶液室温封闭3 h,分别加入鼠源单克隆抗DNAH2或抗β-actin一抗 (1∶1 000),4 ℃孵育过夜。TBST溶液洗膜3次,10 min/次,加入辣根过氧化物酶标记的抗鼠源IgG二抗 (1∶15 000) 室温孵育1 h,TBST溶液再次洗膜3次,10 min/次。加入BeyoECL Plus (超敏ECL化学发光试剂盒)后暗室曝光。
1.2.7 PCR-基因测序分析
常规培养单克隆细胞株,提取基因组DNA,以PCR法扩增包含第1个外显子的DNA片段,再以DNA快速凝胶回收试剂盒回收PCR产物后送测序,分析第1个外显子区的序列特点。相关引物序列见表2。
2 结果与分析
2.1 pX462-DNAH2-sgRNA的构建
首先提取pX462质粒 (图谱见图1A),并以BbsⅠ限制性内切酶进行酶切处理,胶回收含BbsⅠ粘性末端的pX462线性片段;由公司合成DNAH2 sgRNA-A、B以及其互补链,退火连接形成含BbsⅠ粘性末端的寡核苷酸双链结构(图1B);然后以T4 DNA连接酶将pX462线性片段分别与DNAH2 sgRNA-A、B寡核苷酸双链结构相连形成pX462-DNAH2-A、pX462-DNAH2-B重组真核表达质粒。将这两种质粒共转染入U2OS细胞中,可表达DNAH2 sgRNA-A、B和Cas9n蛋白。在sgRNA引导下,Cas9n蛋白识别DNAH2基因的第1个外显子进行剪切、重组,最终实现DNAH2基因的敲除 (图1C)。

表2 PCR及基因测序引物序列Table 2 The primer sequences of the PCR and gene sequencing

图1 pX462载体及sgRNA寡核苷酸双链序列Fig. 1 The enzyme digestion of pX462-DNAH2-sgRNA recombinant plasmid. (A) The map of pX462 plasmid. (B) The A, B sgRNAs and complementary strands were designed and synthesized. The double-stranded oligonucleotide structures containingBbsⅠ cohesive ends were formed during annealing. (C) The constructed recombinant eukaryotic expression plasmids, including pX462-DNAH2-A, pX462-DNAH2-B, can lead to the knocking out ofDNAH2gene, through the role of two sgRNAs in the exon 1 regions ofDNAH2gene.
2.2 pX462-DNAH2-sgRNA的酶切与测序鉴定
分别以BbsⅠ限制性内切酶酶切与基因测序方法对重组真核表达质粒pX462-DNAH2-A、pX462-DNAH2-B进行鉴定。实验结果如图2所示,pX462空载质粒可被BbsⅠ内切酶酶切为线性载体 (泳道2);但成功插入DNAH2 sgRNA-A、B寡核苷酸双链结构的真核表达质粒pX462-DNAH2-A、pX462-DNAH2-B,已经破坏了BbsⅠ酶切位点,故不会被BbsⅠ酶切割为线性质粒 (泳道4,6)。同时,基因测序结果(图3) 表明DNAH2 sgRNA-A、B寡核苷酸双链已成功插入pX462载体。
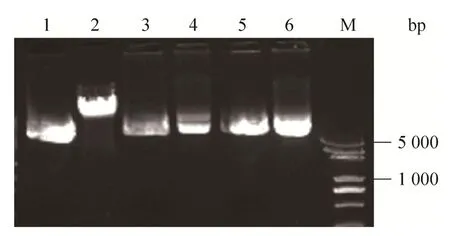
图2 pX462-DNAH2-sgRNA重组质粒的酶切鉴定Fig. 2 The enzyme digestion of pX462-DNAH2-sgRNA recombinant plasmid. 1: pX462 vector/BbsⅠ (-); 2: pX462 vector/BbsⅠ (+); 3: pX462-DNAH2- sgRNA-A/BbsⅠ (-); 4: pX462-DNAH2-sgRNA-A/BbsⅠ (+); 5: pX462-DNAH2-sgRNA-B/BbsⅠ (-); 6: pX462-DNAH2-sgRNAB/BbsⅠ (+); M: marker.

图3 pX462-DNAH2-sgRNA重组质粒的基因测序鉴定Fig. 3 The gene sequencing of pX462-DNAH2-sgRNA recombinant plasmid.
2.3 U2OS-DNAH2-KO单克隆细胞稳定株的蛋白印迹鉴定
通过筛选,获得U2OS-DNAH2-KO的3个单克隆株 (#1−#3) 和U2OS-DNAH2-WT,再分别以NP-40裂解液裂解细胞获全细胞裂解液,以鼠源抗DNAH2、β-actin抗体进行蛋白印迹鉴定。实验结果如图4所示,U2OS-DNAH2-WT细胞 (泳道1) 和U2OS-DNAH2-KO细胞株的#2号 (泳道3) 表达DNAH2蛋白,而U2OS-DNAH2-KO细胞株的#1号 (泳道2) 与#3号 (泳道4) 中DNAH2不表达。β-actin内参蛋白同时表达于以上4个细胞株。这些证明#1、#3这两个细胞株中的DNAH2基因已被敲除。
2.4 U2OS-DNAH2-KO单克隆细胞稳定株的PCR-基因测序分析
分别对U2OS-DNAH2-WT细胞和U2OSDNAH2-KO细胞株 (#1、#3) 进行PCR-基因测序分析。结果如图5所示,U2OS-DNAH2-WT单克隆细胞稳定株没有发生基因突变;而U2OS-DNAH2-KO (#1) 细胞株中在sgRNA (A)与sgRNA (B) 识别区域间发现多处点突变,以及8个碱基的插入突变;U2OS-DNAH2-KO (#3)细胞株也发生类似多处点突变和1个碱基的插入突变。两者均可导致DNAH2基因发生移码突变。

图4 U2OS-DNAH2-KO细胞稳定株的蛋白印迹实验Fig. 4 Western blotting of U2OS-DNAH2-KO stable cell lines. 1: U2OS-DNAH2-WT; 2: U2OS-DNAH2-KO (#1); 3: U2OS-DNAH2-KO (#2); 4: U2OS-DNAH2-KO (#3).

图5 单克隆细胞株的PCR-基因测序分析Fig. 5 PCR-gene sequencing analysis of monoclonal cell lines. (A) U2OS-DNAH2-WT. (B) U2OS-DNAH2-KO (#1). (C) U2OS-DNAH2-KO (#3). “CCC”: the complementary “NGG” site for sgRNA-A; “AGG”: the “NGG” site for sgRNA-B; left black line: binding sequence of sgRNA-A; right black line: complementary binding sequence of sgRNA-B; black arrows: point mutation; red dot: insertion mutation.
3 讨论
利用siRNA (Small interfering RNA)、shRNA (Small hairpin RNA) 等技术可以下调细胞内目的基因的表达,但这种技术不能完全敲除细胞内的目的基因。基因编辑技术则是利用多种基因修饰手段从DNA水平对目的基因的断裂区进行缺失、插入、替换和修复等靶向改造,最终可以导致细胞或动物体内目的基因的完全敲除[9-12]。目前,最常见的基因编辑系统包括锌指核酸酶(Zinc finger nucleases,ZFN)、转录激活因子样效应物核酸酶 (Transcription activator-like effector nucleases,TALEN) 和CRISPR/Cas9[13]。ZFN系统是由FokⅠ核酸内切酶和一系列锌指蛋白组成,两个ZFN分别与目的基因特定位点识别后激活FokⅠ,后者介导DNA的双链断裂[14]。TALEN系统是由FokⅠ酶和TALE蛋白组成,利用TALE的序列模块组装成模块化组合蛋白,后者可与DNA序列结合,激活FokⅠ酶的双链切割功能[15]。CRISPR/Cas9系统则是由Cas9核酸内切酶和CRISPR相关基因组成,基本原理是首先在前间区序列邻近基序 (Protospacer adjacent motif,PAM) 处人工设计sgRNA,后者可特异性结合于目的基因靶序列,形成杂合双链;Cas9酶在sgRNA的引导下,能够对双链进行切割,造成DNA的双链断裂;细胞通过错误倾向的非同源末端连接、同源重组修复等机制对切断的双链进行修复,在断裂处插入异常碱基或致使正常碱基缺失,造成移码突变,最终实现目的基因的敲除[16-18]。作为第3代基因组定点编辑技术,CRISPR/Cas9系统可操作性强,已广泛用于人、细菌、病毒、动物及植物的基因编辑研究,在多种基因敲除或标签标记稳定细胞株的构建、不同基因敲除或转基因动物模型的建立和致病突变基因的校正等方面发挥重要作用[19-21]。
CRISPR/Cas9系统基因编辑的高效性有赖于sgRNA与目的基因靶序列的特异性碱基配对;错配的出现会导致脱靶效应 (Off-target effects) 的产生[19,22]。学者们常通过软件分析、特定实验设计来评估或尽量排除脱靶效应的影响,比如同时采用同一目的基因不同区域的多个sgRNAs进行实验,观察是否出现相同表型,或通过质粒转染方式使基因敲除细胞株重新表达目的基因,观察敲除基因的表型是否恢复。另外,也可通过多种手段或方法从技术层面来降低脱靶效应,比如设计与非靶标位点互补性最低的高效sgRNA序列;改造或寻找更强效的Cas酶;采用确保靶向活性的最低质粒浓度以及针对动物模型的多次交配稀释法[22-25]。CRISPR/Cas9系统已得到不同程度的改进及应用,本实验所采用的CRISPR/Cas9n double nick改良系统有助于减少脱靶效应的影响[10,22,26-29]。野生型Cas9酶会对DNA双链进行切割,而CRISPR/Cas9n double nick系统采用的是Cas9酶的突变体 (Cas9 nickase,Cas9n),其只对与sgRNA结合的DNA单链进行切割,后者容易被细胞修复。因此,只有当两个距离相近的一对sgRNA(A/B) 同时识别并结合目的基因的特定区域后,Cas9n酶才可通过两个相邻的单链切口来实现DNA双链的断裂,从而降低了脱靶效应。
DNAH2基因定位于17p13[3],可编码DNAH2蛋白,后者包括4 427个氨基酸,相对分子量约为500 kDa (NP_065928.2)。研究发现,DNAH2蛋白中的P-loop-NTPase结构域含有经典的核苷酸磷酸盐结合模序 (Walker A与Walker B模序),并可通过三磷酸核苷水解酶的作用释放能量,介导微管间的相互移动,最终参与纤毛的运动[1-3]。目前,学者对于DNAH2基因的研究数据非常有限。为了方便探讨DNAH2基因的功能,本实验利用CRISPR/Cas9n系统建立了DNAH2基因靶向敲除U2OS细胞株。另外,考虑到范可尼贫血的相关突变基因多编码与DNA损伤修复相关的蛋白[5],利用DNAH2基因敲除细胞株,从DNA损伤修复角度深入分析DNAH2基因在范可尼贫血中的作用机制是本实验室以后工作的重点。
REFERENCES
[1] Jones RT, Abedalthagafi MS, Brahmandam M, et al. Cross-reactivity of the BRAF VE1 antibody with epitopes in axonemal dyneins leads to staining of cilia. Mod Pathol, 2015, 28(4): 596–606.
[2] Chapelin C, Duriez B, Magnino F, et al. Isolation of several human axonemal dynein heavy chain genes: genomic structure of the catalytic site, phylogenetic analysis and chromosomal assignment. FEBS Lett, 1997, 412(2): 325–330.
[3] Pazour GJ, Agrin N, Walker BL, et al. Identification of predicted human outer dynein arm genes: candidates for primary ciliary dyskinesia genes. J Med Genet, 2006, 43(1): 62–73.
[4] Morillas HN, Zariwala M, Knowles MR. Genetic causes of bronchiectasis: primary ciliary dyskinesia. Respiration, 2007, 74(3): 252–263.
[5] Duxin JP, Walter JC. What is the DNA repair defect underlying Fanconi anemia?. Curr Opin Cell Biol, 2015, 37: 49–60.
[6] Bogliolo M, Surrallés J. Fanconi anemia: a model disease for studies on human genetics and advanced therapeutics. Curr Opin Genet Dev, 2015, 33: 32–40.
[7] Longerich S, Li J, Xiong Y, et al. Stress and DNA repair biology of the Fanconi anemia pathway. Blood, 2014, 124(18): 2812–2819.
[8] Chang LX, Yuan WP, Zeng HM, et al. Whole exome sequencing reveals concomitant mutations of multiple FA genes in individual Fanconi anemia patients. BMC Med Genomics, 2014, 7: 24.
[9] Boettcher M, McManus MT. Choosing the right tool for the job: RNAi, TALEN, or CRISPR. Mol Cell, 2015, 58(4): 575–585.
[10] Ran FA, Hsu PD, Wright J, et al. Genome engineering using the CRISPR-Cas9 system. Nat Protoc, 2013, 8(11): 2281–2308.
[11] Yang H, Wang HY, Shivalila CS, et al. One-step generation of mice carrying reporter and conditional alleles by CRISPR/Cas-mediated genome engineering. Cell, 2013, 154(6): 1370–1379.
[12] Ma YW, Ma J, Zhang X, et al. Generation ofeGFPandCreknockin rats by CRISPR/Cas9. FEBS J, 2014, 281(17): 3779–3790.
[13] Doudna JA, Charpentier E. Genome editing. The new frontier of genome engineering with CRISPR-Cas9. Science, 2014, 346(6213): 1258096.
[14] Ul Ain Q, Chung JY, Kim YH. Current and future delivery systems for engineered nucleases: ZFN, TALEN and RGEN. J Control Release, 2015, 205: 120–127.
[15] Wright DA, Li T, Yang B, et al. TALEN-mediated genome editing: prospects and perspectives. Biochem J, 2014, 462(1): 15–24.
[16] Barrangou R. RNA events. Cas9 targeting and the CRISPR revolution. Science, 2014, 344(6185): 707–708.
[17] Cong L, Ran FA, Cox D, et al. Multiplex genome engineering using CRISPR/Cas systems. Science, 2013, 339(6121): 819–823.
[18] Mali P, Yang LH, Esvelt KM, et al. RNA-guided human genome engineeringviaCas9. Science, 2013, 339(6121): 823–826.
[19] Duan JZ, Lu GQ, Xie Z, et al. Genome-wide identification of CRISPR/Cas9 off-targets in human genome. Cell Res, 2014, 24(8): 1009–1012.
[20] Jiang WY, Bikard D, Cox D, et al. RNA-guidedediting of bacterial genomes using CRISPR-Cas systems. Nat Biotechnol, 2013, 31(3): 233–239.
[21] Kennedy EM, Kornepati AVR, Mefferd AL, et al. Optimization of a multiplex CRISPR/Cas system for use as an antiviral therapeutic. Methods, 2015, 91: 82–86.
[22] Wu Z, Feng G. Progress of application and off-target effects of CRISPR/Cas9. Hereditas, 2015, 37(10): 1003–1010.
[23] Ran FA, Cong L, Yan WX, et al. In vivo genome editing using Staphylococcus aureus Cas9. Nature, 2015, 520(7546): 186–191.
[24] Hsu PD, Lander ES, Zhang F. Development and applications of CRISPR-Cas9 for genome engineering. Cell, 2014, 157(6): 1262–1278.
[25] Shengsong X, Yi Z, Lisheng Z, et al. sgRNA design for the CRISPR/Cas9 system and evaluation of its off-target effects. Hereditas, 2015, 37(11): 1125–1136.
[26] Ran FA, Hsu PD, Lin CY, et al. Double nicking by RNA-guided CRISPR Cas9 for enhanced genome editing specificity. Cell, 2013, 154(6): 1380–1389.
[27] Shen B, Zhang WS, Zhang J, et al. Efficient genome modification by CRISPR-Cas9 nickase with minimal off-target effects. Nat Methods, 2014, 11(4): 399–402.
[28] Fu YF, Sander JD, Reyon D, et al. Improving CRISPR-Cas nuclease specificity using truncated guide RNAs. Nat Biotechnol, 2014, 32(3): 279–284.
[29] Trevino AE, Zhang F. Genome editing using Cas9 nickases. Methods Enzymol, 2014, 546: 161–174.
(本文责编 陈宏宇)
Knocking out of humanDNAH2gene in U2OS cells by CRISPR/Cas9n double nick system
Lixian Chang, Congcong Sun, Xiaojuan Chen, Wenyu Yang, Jiayuan Zhang, Yingchi Zhang, Weiping Yuan, and Xiaofan Zhu
Institute of Hematology and Blood Diseases,Hospital Chinese Academy of Medical Sciences & Peking Union Medical College,Tianjin300020,China
To study the biological function ofDNAH2(Homo sapiens dynein, axonemal, heavy chain 2) gene, we constructed human stable U2OS cell line of DNAH2 gene knockout through CRISPR/Cas9n double nick system. The A, B sgRNAs (Single guide RNA) and complementary strands were designed and synthesized. The double-stranded structures were formed during annealing, and connected with BbsⅠ cohesive ends-containing pX462 linear vector to construct the recombinant eukaryotic expression plasmids, including pX462-DNAH2-A and pX462-DNAH2-B. After the co-transfection of the two plasmids into U2OS cells, the addition of puromycin and limiting dilution method were used to obtain positive monoclonal cell line. Western blotting assay was then performed to detect the expression of DNAH2 protein, and PCR-sequencing technology was finally utilized to analyze the mutation feature. The results showed that A, B sgRNAs duplex was successfully inserted into pX462 vector, and DNAH2 protein was not expressed andDNAH2gene suffered from the frame-shift mutation in U2OS-DNAH2-KO monoclonal cell line. These demonstrated that DNAH2 knockout U2OS stable cell line was successfully constructed through CRISPR/Cas9n double nick system, which providing a useful tool for the study of DNAH2 gene.
DNAH2, CRISPR/Cas9n, knock-out, stable cell line
Xiaofan Zhu. Tel/Fax: +86-22-23909196; E-mail: xfzhu@ihcams.ac.cn
Received:July 21, 2016;Accepted:September 26, 2016
Supported by: National Natural Science Foundation of China (Nos. 81500156, 81470339, 81470280, 81300394), Tianjin Science and Technology Project (No.12ZCDZSY18100).
国家自然科学基金 (Nos. 81500156, 81470339, 81470280, 81300394),天津市科技计划项目 (No. 12ZCDZSY18100) 资助。
