UPLC-MS/MS法测定人血浆中罗匹尼罗的浓度及其药动学研究Δ
2017-02-23黄晨蓉许青青缪丽燕苏州大学附属第一医院临床药理室江苏苏州15006苏州大学药学院江苏苏州15000
黄晨蓉,许青青,缪丽燕(1.苏州大学附属第一医院临床药理室,江苏苏州 15006;.苏州大学药学院,江苏苏州 15000)
UPLC-MS/MS法测定人血浆中罗匹尼罗的浓度及其药动学研究Δ
黄晨蓉1,2*,许青青2,缪丽燕1#(1.苏州大学附属第一医院临床药理室,江苏苏州 215006;2.苏州大学药学院,江苏苏州 215000)
目的:建立测定人血浆中罗匹尼罗浓度的方法,并用于药动学研究。方法:血浆样品经乙腈沉淀后,以阿替洛尔为内标,采用超高效液相色谱-串联质谱法测定。色谱柱为WatersACQUITY UPLC BEHAmide,流动相为水(含10 mmol/L乙酸铵和0.1%甲酸)-乙腈(85∶15,V/V),流速为0.3 mL/min,柱温为40℃,进样量为5 μL。采用电喷雾离子源,以多反应监测方式进行正离子扫描,用于定量分析的离子对分别为m/z 261.2→114.1(罗匹尼罗)和m/z 267.2→145.0(内标)。选择8例健康受试者,男、女各半,单次给予盐酸罗匹尼罗片1.0 mg后,采用该法测定给药前后罗匹尼罗的血药浓度,采用WinNonlin 6.3软件计算其药动学参数。结果:罗匹尼罗血药浓度在0.02~2 ng/mL范围内线性关系良好(r=0.997 3),批内、批间RSD<10%,准确度为95.2%~99.7%,提取回收率为68.5%~79.9%,基质效应和稀释效应均不影响其血药浓度的测定。8例健康受试者口服盐酸罗匹尼罗片1.0 mg后,cmax为(2.1±0.5)ng/mL,tmax为(1.0±0.5)h,t1/2为(4.7±1.5)h,AUC0-36h为(14.7±6.0)ng·h/mL,AUC0-∞为(15.1±6.1)ng·h/mL;不同性别受试者主要药动学参数比较,差异均无统计学意义(P>0.05)。结论:该方法操作简便、灵敏度高、分析时间短,适用于人血浆中罗匹尼罗的浓度测定及药动学研究。
超高效液相色谱-串联质谱法;罗匹尼罗;血药浓度;药动学
盐酸罗匹尼罗(Ropinirole hydrochloride)为非麦角碱类多巴胺受体激动药,于1996年在英国上市,现已在多个国家临床应用于治疗帕金森病(Parkinson disease,PD)[1]。与传统多巴胺受体激动药相比,盐酸罗匹尼罗避免了因麦角结构引起的严重纤维化不良反应;同时,由于其与受体的高度选择性,还可降低左旋多巴所致运动障碍的发生率;此外,盐酸罗匹尼罗还具有神经保护作用[2]。临床研究显示,盐酸罗匹尼罗不论是单药治疗早期PD还是联合左旋多巴治疗晚期PD,均表现出较好的疗效;且PD患者早期接受盐酸罗匹尼罗的单药治疗还可推迟联用左旋多巴的时间或减少其使用剂量,延迟PD并发症的发生,长期使用安全、有效,且耐受性较好[3-4]。目前,国内外研究多采用高效液相色谱-串联质谱(LC-MS/MS)法测定人血浆中罗匹尼罗的浓度,以乙酸乙酯、甲基叔丁醚等为溶剂,采用液-液萃取法处理血浆样品,操作烦琐且耗时[5-11]。故本试验在此基础上,建立了一种快速、灵敏的超高效液相色谱-串联质谱(UPLC-MS/MS)法,并将其应用于临床药动学研究,为盐酸罗匹尼罗Ⅱ期临床试验方案的制订提供参考。
1 材料
1.1 仪器
Acquity UPLC仪、Xevo-TQS三重四级杆串联质谱仪(美国Waters公司);XS105DU型分析天平(美国Mettler公司);Allegra 64R型高速冷冻离心机(美国Beckman公司);Milli-Q型超纯水仪(美国Millipore公司)。
1.2 药品与试剂
盐酸罗匹尼罗对照品(美国Pharmacopeia公司,批号:F0J036,纯度:99.5%);阿替洛尔对照品(内标,中国食品药品检定研究院,批号:100117-201105,纯度:99.8%);盐酸罗匹尼罗片(浙江华海药业股份有限公司,批号:11813001,规格:1.0 mg/片);甲醇、乙腈、甲酸为色谱纯,其余试剂均为分析纯,水为纯化水。
2 方法与结果
2.1 色谱与质谱条件
色谱柱:Waters ACQUITY UPLC BEH Amide(50 mm×2.1 mm,1.7 μm);流动相:水(含10 mmol/L乙酸铵和0.1%甲酸)-乙腈(85∶15,V/V);流速:0.3 mL/min;柱温:40℃;进样量:5 μL。
采用电喷雾离子源(Electrospray ionization,ESI),以多反应监测(Multiple reaction monitoring,MRM)模式扫描,正离子方式检测。毛细管电压:1.34 kV;锥孔电压:70 V;碰撞电压:20 eV;脱溶剂温度:500℃;脱溶剂气流速:1 000 L/h;锥孔气流速:150 L/h。用于定量分析的离子对分别为:m/z 261.2→114.1(罗匹尼罗)和m/z 267.2→145.0(内标)。
2.2 溶液的制备
精密称取盐酸罗匹尼罗对照品11.40 mg(相当于罗匹尼罗9.998 mg),置于10 mL量瓶中,用甲醇溶解并定容,得罗匹尼罗质量浓度为1 mg/mL的贮备液,置于4℃冰箱中保存。用50%乙腈稀释成相应质量浓度的标准工作液,备用。
另精密称取内标对照品10.26 mg,置于10 mL量瓶中,用甲醇溶解并定容,得质量浓度为1.026 mg/mL的内标贮备液;取上述内标贮备液适量,用50%乙腈稀释成质量浓度为500 ng/mL的内标溶液,置于4℃冰箱中保存,备用。
2.3 血浆样品处理
精密吸取空白血浆0.1 mL,加入50%乙腈10 μL,加入内标溶液(500 ng/mL)10 μL,加入乙腈0.1 mL,涡旋振荡1 min,在4℃下,以19 722×g离心10 min,取上清液100 μL至进样瓶中,进样分析。
2.4 方法学验证
2.4.1 专属性考察 在“2.1”项条件下,内源性杂质对待测物无干扰,罗匹尼罗和内标的峰形较好,分离完全,保留时间分别约为1.2和1.0 min。典型色谱图见图1。

图1 典型MRM色谱图Fig 1 Typical MRM chromatograms
2.4.2 标准曲线的绘制与定量下限的考察 精密吸取空白血浆、罗匹尼罗标准工作液各适量,配制成质量浓度分别为0.02、0.05、0.1、0.2、0.5、1、2 ng/mL的血浆样品,按“2.3”项下方法处理后,进样测定,记录色谱图。以待测物质量浓度(c)为横坐标、待测物与内标的峰面积比值(f)为纵坐标,采用加权最小二乘法(权重系数w=1/c2)进行线性回归,得回归方程为f=31.239 7c+50.890 5(r=0.997 3)。结果显示,罗匹尼罗的血药浓度在0.02~2 ng/mL范围内线性关系良好,其定量下限为0.02 ng/mL(批内RSD<20%)。
2.4.3 精密度与准确度试验 分别配制罗匹尼罗低、中、高质量浓度(0.05、0.2、1.5 ng/mL,下同)的质控(QC)样品,按“2.3”项下方法处理,每质量浓度取6样本分析,连续测定3批,根据当批标准曲线计算各样品的测得质量浓度,考察方法的精密度与准确度,结果见表1。
表1 精密度与准确度试验结果()Tab 1 Results of precision and accuracy tests()
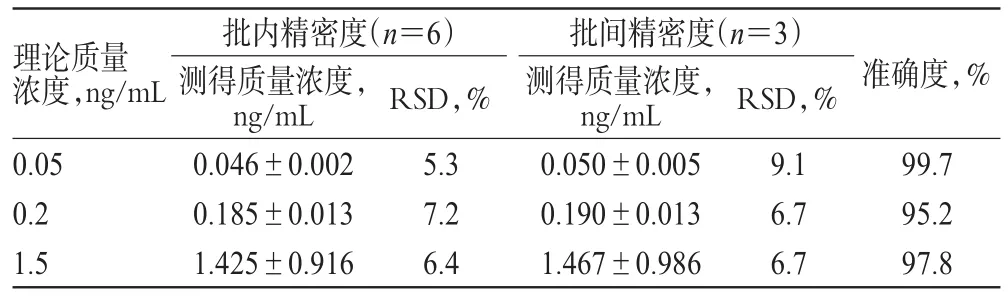
表1 精密度与准确度试验结果()Tab 1 Results of precision and accuracy tests()
理论质量浓度,ng/mL准确度,% 0.05 0.2 1.5批内精密度(n=6)测得质量浓度,ng/mL 0.046±0.002 0.185±0.013 1.425±0.916 RSD,% 5.3 7.2 6.4批间精密度(n=3)测得质量浓度,ng/mL 0.050±0.005 0.190±0.013 1.467±0.986 RSD,% 9.1 6.7 6.7 99.7 95.2 97.8
由表1可见,各质量浓度QC样品的批内RSD分别为5.3%、7.2%和6.4%,批间RSD分别为9.1%、6.7%和6.7%,准确度为95.2%~99.7%,精密度与准确度均符合相关指导原则的基本要求[12]。
2.4.4 提取回收率试验 分别配制罗匹尼罗低、中、高质量浓度的QC样品各6份,按“2.3”项下方法处理后,进样测定,得相应色谱峰峰面积(A1);取空白血浆适量,按“2.3”项下方法处理后,加入相应质量浓度的罗匹尼罗标准工作液,使最终质量浓度与前者相对应,进样测定,得相应色谱峰峰面积(A2)。提取回收率(%)=A1/A2× 100%。结果显示,各质量浓度QC样品罗匹尼罗的提取回收率分别为68.5%、77.7%和79.9%;内标的提取回收率为68.3%,RSD<15%(n=6)。
2.4.5 基质效应 取空白血浆适量,按“2.3”项下方法处理后,加入相应质量浓度的罗匹尼罗标准工作液和内标溶液适量,进样测定,得相应色谱峰峰面积(C);另取同体积相应质量浓度的标准工作液和内标溶液,以氮气流吹干,残渣用流动相复溶,使最终质量浓度与前者相对应,进样测定,得相应色谱峰峰面积(B)。基质效应(%)=C/B×100%。结果显示,各质量浓度QC样品罗匹尼罗的基质效应分别为81.1%、92.1%和94.3%,内标的基质效应为93.0%,表明基质效应不影响罗匹尼罗血药浓度的测定[12]。
2.4.6 稀释效应 配制质量浓度为3 ng/mL的血浆样品共6份,用空白血浆以体积比1∶1的比例稀释后,按“2.3”项下方法处理,进样测定,考察各样品的稀释可靠性,计算各样品测得质量浓度与理论质量浓度的偏差。结果显示,各样品的偏差为7.2%~12.7%,符合相关指导原则基本要求[12]。
2.4.7 稳定性试验 配制罗匹尼罗低、中、高质量浓度的QC样品,分别于室温放置6 h、经历3次冻融(每次冷冻保存的时间>12 h)、-80℃放置45 d后,按“2.3”项下方法处理,进样测定,考察各样品的稳定性;同时考察各样品按“2.3”项下方法处理后于自动进样器(10℃)中放置24 h的稳定性。计算各样品的测得质量浓度与理论质量浓度的偏差。结果显示,各样品的偏差为-9.8%~11.7%,RSD<15%,表明其在上述条件下稳定性良好。
2.4.8 质量控制 分别配制罗匹尼罗低、中、高质量浓度的QC样品,与每个分析批的待测样品同时测定,计算各QC样品测得质量浓度与理论质量浓度的偏差。结果显示,各批QC样品测定结果的偏差均<15%,符合生物样品定量分析的基本要求[12]。
2.5 药动学研究
2.5.1 研究对象 选择8例健康受试者,男性、女性各4例,平均年龄(25.5±1.7)岁,平均身高(1.7±0.1)m,平均体质量(60.3±7.4)kg,平均体质量指数(21.4±1.9)kg/m2。受试者试验前心电图、血压、血尿常规、肝肾功能检查均正常,无药物过敏史,无慢性疾病史;试验前1个月及试验期间未使用过其他药物。受试期间统一清淡饮食,忌烟、酒及含药物、咖啡因的饮料,不食用影响肝药酶的水果及饮料。本试验方案经苏州大学附属第一医院医学伦理委员会审核批准,受试者均知情同意并签署知情同意书。
2.5.2 单剂量药动学研究 本试验采用开放、单中心、单剂量临床试验设计,给药剂量为1.0 mg。受试者隔夜禁食,于次日晨起空腹口服盐酸罗匹尼罗片1.0 mg,以200 mL温水送服,给药后2 h内不饮水,4 h后统一进食标准餐。分别于给药前及给药后0.5、0.75、1.0、1.5、2.0、2.5、3.0、4.0、6.0、8.0、12.0、24.0和36.0 h采集前臂静脉血4 mL,置肝素抗凝管中,以2 352×g离心10 min,分离血浆,置于-80℃冰箱中冷冻保存,待测。
采用UPLC-MS/MS法测定不同时间点受试者体内罗匹尼罗的血药浓度。若血药浓度超过了线性范围上限,则将样品用空白血浆以体积比1∶1的比例稀释,按“2.3”项下方法处理后,重新测定。采用WinNonlin 6.3软件计算药动学参数,采用SPSS 16.0软件处理所得数据,计量资料以表示,组间比较采用t检验。P<0.05为差异有统计学意义
8例健康受试者单剂量口服盐酸罗匹尼罗片后的平均药-时曲线见图2,主要药动学参数见表2。

图2 8例健康受试者单剂量口服盐酸罗匹尼罗片后的平均药-时曲线(n=8)Fig 2 Mean plasma concentration-time curve in 8 healthy volunteers after a single oral dose of Ropinirole hydrochloride tablets(n=8)
表2 主要药动学参数()Tab 2 Main pharmacokinetic parameters()
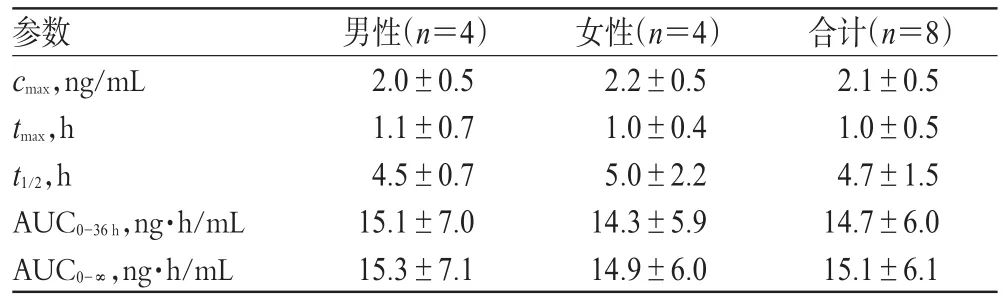
表2 主要药动学参数()Tab 2 Main pharmacokinetic parameters()
参数cmax,ng/mL tmax,h t1/2,h AUC0-36h,ng·h/mL AUC0-∞,ng·h/mL男性(n=4)2.0±0.5 1.1±0.7 4.5±0.7 15.1±7.0 15.3±7.1女性(n=4)2.2±0.5 1.0±0.4 5.0±2.2 14.3±5.9 14.9±6.0合计(n=8)2.1±0.5 1.0±0.5 4.7±1.5 14.7±6.0 15.1±6.1
由表2可见,男性受试者和女性受试者主要药动学参数比较,差异均无统计学意义(P>0.05),提示性别对其主要药动学参数无明显影响。
8例受试者中,有7例受试者发生药品不良事件,其中恶心6例、头晕4例、呕吐1例、多汗2例,均发生在给药后40~60 min,其中1例受试者在给药后142 min时再次出现头晕,并在70 min内自行痊愈。1例受试者出现尿白细胞升高,经医师诊断为异常,具有临床意义,6 d后恢复正常。未见其他严重不良事件发生。
3 讨论
在色谱条件优化过程中,笔者考察了Waters ACQUITY UPLC BEHAmide(50 mm×2.1 mm,1.7 μm)及不同规格的Waters ACQUITY UPLC BEH C18(50 mm×2.1 mm,1.7 μm和100 mm×2.1 mm,1.7 μm)等色谱柱。结果显示,相比于BEH C18,BEH Amide可得到更好的色谱峰形。在选择流动相时,笔者考察了0.1%甲酸水溶液、10 mmol/L乙酸铵水溶液和水(含10 mmol/L乙酸铵和0.1%甲酸)对检测的影响。结果显示,后者可得到较好的峰形,且响应值最大,故选择其作为水相。
在质谱条件优化过程中,对罗匹尼罗母离子和子离子进行扫描,发现正离子模式响应强,故采用正离子检测模式。在质谱参数的优化过程中,根据信号强度将罗匹尼罗的定量离子对确定为m/z 261.2→114.1,锥孔电压和碰撞电压分别确定为70 V和20 eV。在相同检测条件下,筛选了多种内标物质(如奥美拉唑、氟氯西林、阿替洛尔等),根据信号强度及与罗匹尼罗色谱峰的分离度,最终确定内标为阿替洛尔,其定量离子对为m/z 267.2→145.0,锥孔电压为70 V,碰撞电压为20 eV。
在血浆样品处理方法的筛选中,笔者尝试了蛋白沉淀法和液-液萃取法,发现蛋白沉淀法可有效去除蛋白;而以乙酸乙酯、甲基叔丁醚等为溶剂的液-液萃取法[8-9],虽然也能有效去除蛋白,但是操作烦琐且耗时,不利于大批量样本处理。因此,本试验最终选择简单易行的蛋白沉淀法,与文献报道的方法[5-11]比较,该法重复性良好,内源性物质干扰较少,可满足生物样本快速批量检测的需求。
对本试验建立的UPLC-MS/MS法进行方法学考察,结果显示,在“2.1”项条件下,罗匹尼罗和内标的保留时间分别约为1.2和1.0 min,峰形良好,不受血浆中内源性物质的干扰;罗匹尼罗的血药浓度在0.02~2 ng/mL范围内线性关系良好,批内、批间RSD<10%;提取回收率、基质效应和稀释效应等均符合生物样品定量分析的基本要求[12],且稳定性良好。
罗匹尼罗是一种非麦角碱类的多巴胺受体激动药,目前国外临床研究多在患者中开展,且根据药物的不用剂型设计不同的给药方案。药品说明书指出,该药普通片剂成人初始剂量为0.25 mg,tid,每周向上调整,即第1周剂量为0.25 mg,tid;第2周剂量为0.5 mg,tid;第3周剂量为0.75 mg,tid;第4周剂量为1.0 mg,tid,剂量最高可增加至3.0 mg,tid。常用维持日剂量为3~9 mg,最大日剂量为24 mg。而以健康受试者为研究对象的临床试验,国内已有文献报道[13],其剂量低于临床常用剂量,为单次口服1.0 mg,未见相关不良事件。因此,本试验将低于临床常用剂量且文献报道安全的1.0 mg设定为给药剂量,开展单剂量、单中心药动学试验。结果显示,8例健康受试者(男、女各半)单次口服盐酸罗匹尼罗1.0 mg后的主要药动学参数为cmax(2.1±0.5)ng/mL、t1/2(4.7±1.5)h、tmax(1.0±0.5)h,且性别对主要药动学参数无明显影响(P>0.05)。试验过程中,有7例受试者发生药品不良事件,包括恶心、头晕、呕吐、多汗、尿白细胞升高等,医师综合判断可能与药物相关,临床应予以重视。
本试验仅初步考察了单次口服盐酸罗匹尼罗片后在健康人体内的药动学特性,在后续试验中,本课题组将进一步开展以PD患者为研究对象的多剂量、多中心药动学研究,以期为盐酸罗匹尼罗片后续临床试验给药方案的制订提供理论依据。
(本研究经原国家食品药品监督管理局批准,临床试验批件号:2012L00005)
[1] 胡尚久,王继生,张润峰.新型多巴胺受体激动剂罗匹尼罗的药代动力学研究进展[J].中国药房,2003,14(3):182-184.
[2] 罗再贵.罗匹尼罗:治疗帕金森病的新型多巴胺激动剂[J].川北医学院学报,2002,17(2):117-119.
[3] Rascol O,Brooks DJ,Korczyn AD,et al.A five-year study of the incidence of dyskinesia in patients with early Parkinson’s disease who were treated with ropinirole or levodopa[J].N Engl J Med,2000,342(20):1484-1491.
[4] Mizuno Y,Abe T,Hasegawa K,et al.Ropinirole is effective on motor function when used as an adjunct to levodopa in Parkinson’s disease:STRONG study[J].Mov Disord,22(13):1860-1865.
[5]Mohamed S,Riva R,Contin M.Validated UPLC-MS/MS method for the simultaneous determination of pramipexole and ropinirole in plasma of patients with Parkinson’s disease[J].J Chromatogr B Analyt Technol Biomed Life Sci,2016,doi:10.1016/j.jchromb.2016.02.041.
[6] Fuster J,Negro S,Salama A,et al.HPLC-UV method development and validation for the quantification of ropinirole in new PLGA multiparticulate systems:microspheres and nanoparticles[J].Int J Pharm,2015,491(1/2):310-317.
[7] Wen AD,Jia YY,Luo XX,et al.The effect of Madopar on the pharmacokinetics of ropinirole in healthy Chinese volunteers[J].J Pharm Biomed Anal,2007,43(2):774-778.
[8] Bharathi DV,Jagadeesh B,Kumar SS,et al.Highly sensitive method for the determination of ropinirole with a lower limit of quantitation of 3.45 pg/mL in human plasma by LC-ESI-MS/MS:application to a clinical pharmacokinetic study[J].Biomed Chromatogr,2009,23(5):557-562.
[9] 王洪允,刘明,胡蓓,等.超高效液相色谱-串联质谱法测定人血浆中罗匹尼罗浓度[J].质谱学报,2015,36(4):296-301.
[10] 张婉直,王秀华,惠民权,等.盐酸罗匹尼罗渗透泵型控释片的药代动力学研究[J].西北药学杂志,2016,31(2):172-176.
[11] 王子腾,黄晨蓉,许青青,等.液相色谱-质谱联用法研究大鼠血浆及纹状体中罗匹尼罗药动学[J].中国新药杂志,2015,24(12):1412-1417、1422.
[12]《化学药物临床药代动力学研究技术指导原则》课题研究组.化学药物临床药代动力学研究技术指导原则[S]. 2005-03-18.
[13] 丁莉坤,杨林,王茂湖,等.LC-MS/MS法测定人血浆中罗匹尼罗及联合多巴丝肼片后的药动学[J].中国新药与临床杂志,2013,32(3):240-244.
Determination of Ropinirole in Human Plasma by UPLC-MS/MS and Its Pharmacokinetic Study
HUANG Chenrong1,2,XU Qingqing2,MIAO Liyan1(1.Dept.of Clinical Pharmacology,the First Affiliated Hospital of Soochow University,Jiangsu Suzhou 215006,China;2.College of Pharmacy,Soochow University,Jiangsu Suzhou 215000,China)
UPLC-MS/MS;Ropinirole;Plasma concentration;Pharmacokinetics
R945
A
1001-0408(2017)02-0177-05
2016-05-20
2016-11-01)
(编辑:张元媛)
国家自然科学基金青年科学基金项目(No.81503159)
*副主任药师,硕士。研究方向:临床药学。电话:0512-67780467。E-mail:chrishuangcr@163.com
#通信作者:主任药师,教授,博士生导师,博士。研究方向:临床药学。电话:0512-67780467。E-mail:miaolysuzhou@163.com
DOI10.6039/j.issn.1001-0408.2017.02.09
ABSTRACTOBJECTIVE:To develop a method for the determination of ropinirole in human plasma and pharmacokinetic study. METHODS:After precipitated with acetonitrile,using atenolol as internal standard,plasma sample was determined by UPLC-MS/ MS.The separation was performed on Waters ACQUITY UPLC BEH Amide column with mobile phase consisted of water(containing 10 mmol/L ammonium acetate and 0.1%formic acid)-acetonitrile(85∶15,V/V)at flow rate of 0.3 mL/min.The column temperature was set at 40℃,and sample size was 5 μL.The analyses were carried out by ESI under MRM model in positive mode.The mass transition ion-pairs were as follows:m/z 261.2→114.1(ropinirole)and m/z 267.2→145.0(internal standard).8 healthy volunteers were selected with a gender ratio of half to half,and were given Ropinirole hydrochloride tablets 1.0 mg.Plasma concentrations of ropinirole were determined before and after medication.The pharmacokinetic parameters were calculated by WinNonlin 6.3 software.RESULTS:The linear range of ropinirole were 0.02-2 ng/mL(r=0.997 3)with RSDs of inter-batch and intra-batch<10%;accuracy ranged 95.2%-99.7%,and extraction recoveries ranged 68.5%-79.9%.Both matrix effects and dilution effects didn’t influence the determination of plasma concentration.The main pharmacokinetic parameters of ropinirole in 8 healthy volunteers after oral administration of Ropinirole hyolrocholride tablets 1.0 mg were as follows:cmaxwas(2.1±0.5)ng/mL,tmaxwas(1.0±0.5)h,t1/2was(4.7±1.5)h,AUC0-36hwas(14.7±6.0)ng·h/mL,AUC0-∞was(15.1±6.1)ng·h/mL.There was no statistical significance in main pharmacokinetic parameters between different gender(P>0.05).CONCLUSIONS:The method is simple,sensitive and less time-consuming.It is suitable for the plasma concentration determination and pharmacokinetic study of ropinirole in human.
