GLP-1R结构和功能及小分子药物筛选研究进展
2017-02-22胡中平程念杨帆苏正定
胡中平 程念 杨帆 苏正定
(湖北工业大学生物医药研究院 工业发酵协同创新中心 教育部发酵工程重点实验室,武汉 430068)
GLP-1R结构和功能及小分子药物筛选研究进展
胡中平 程念 杨帆 苏正定
(湖北工业大学生物医药研究院 工业发酵协同创新中心 教育部发酵工程重点实验室,武汉 430068)
胰高血糖素样肽-1受体(glucagon-like peptide-1 receptor,GLP-1R)作为2-型糖尿病(T2DM)药物研发和治疗的靶点有着十分重要的临床意义。尽管通过结构生物学,蛋白质工程等方法和手段对于GLP-1R结构的研究有了较大突破。但是关于其全长结构解析,多肽结合受体的分子机理及受体激活的内在机制还不曾得到解决。近些年有关GLP-1R相关研究发展较快,简述了该受体的结构与功能以及已有的小分子药物先导化合物,并讨论GLP-1受体分子结构作用机制的发展方向及应用前景,旨为进一步探寻2型糖尿病的治疗方案提供有利的帮助。
GLP-1R;分子结构;小分子药物
胰高血糖素样肽-1受体(glucagon-like peptide-1 receptor,GLP-1R)是2-型糖尿病最为有效的治疗靶点之一。经过多年基础研究积累,转化和临床研究表明GLP-1与其受体相互作用能够有效调控机体糖稳态和能量代谢[1]。GLP-1R属于G蛋白偶联受体(GPCR)B簇亚族(B1)的一员,它的典型特征是具有一个相对比较大的胞外域(ECD)和有α-螺旋束构成的7次跨膜核心域(TMD)。GLP-1R作为GLP-1/GLP-1R途径下游信号的靶标,主要是通过“two-domain model”来激活受体。首先GLP-1 C端域(cGLP-1)同GLP-1R胞外域(ECD)形成的“affinity trap”结合,从而确保GLP-1 N端域(nGLP-1)与受体核心域(TMD)形成的“pocket”互交。这种相互作用能够有效激活PKA、PI3K、MAPK等多种下游信号通路,参与诸如胰岛素的释放,β-细胞增生,胰高血糖素释放减少,延迟胃排空[2],增强记忆等重要生理过程[3]。所以,GLP-1R作为GLP-1发挥效能的靶点对于人们研究多种疾病有着潜在的指导意义。本文就GLP-1R的结构和功能的研究进展进行综述,并针对基于该受体的小分子先导化合物进行归纳,以便为进一步开发合适治疗T2DM的口服药物提供借鉴。
1 GLP-1R的结构特性概况
胰高血糖素样肽(GLP-1)和葡萄糖依赖性促胰岛素多肽(GIP)作为肠促胰素都能够直接作用于胰岛细胞加强餐后胰岛素释放[4,5],但是在治疗2-型糖尿病(T2DM)方面首选GLP-1。主要有两个原因:首先,GLP-1和GIP都会抑制胃的排空,延迟餐后血糖升高,但只有GLP-1会引起饱足感;其次,这两种多肽对胰高血糖素的释放具有相反的效用,GLP-1起抑制作用但GIP会促进胰高血糖素释放[6]。GLP-1主要是由胰高血糖素原基因表达通过肠L细胞分泌的多肽类激素,其很容易被DPP-Ⅳ降解成无生理活性GLP-1(9-37),半衰期较短仅有1-2 min[6]。GLP-1发挥生理功能主要是通过结合并激活G蛋白偶联受体B家族(分泌素家族)中的GLP-1R,诱发信号分子cAMP增加,使偶联的G蛋白α亚基与β,γ亚基解离,并分别介导胞内不同信号通路来完成的[7]。
1.1 GLP-1R的表达及糖基化
人的GLP-1受体(hGLP-1R)基因位于人染色体6p21上,编码463个氨基酸。GLP-1R表达于模式生物和人体中并呈现出高度保守性,它在机体中广泛分布于被检测的胰岛、胃、小肠、心脏、肾脏、肺及大脑等组织中。GLP-1R在胰岛β细胞中表达较多,在人胰岛α和δ细胞中表达尚属争议[8]。人GLP-1受体(hGLP-1R)同小鼠GLP-1受体(rGLP-1R)序列具有高度相似性,同源性达到84%。该受体属于G蛋白偶联受体B簇中胰高血糖素受体亚家族,这类受体有3个显著的特征:一个相对较长的大约100-150个氨基酸的胞外N端域(ECD),与之相连的7次跨膜结构域(7TMD)以及连接跨膜段的相对较短的胞内域C端域(ICD)[9]。其中胞外域(ECD)包括两组通过loop环相连的反向β折叠,6个保守的半胱氨酸形成的3对二硫键以及一个柔性的α螺旋(图1,参照PDB ID:2QKH[10])。研究发现在RINm5F细胞[11]中,糖基化肽酶F能够使GLP-1受体分子量从63 kD减少到51 kD,表明GLP-1受体的N端连接有聚糖分子。衣霉素也会减少GLP-1受体的表达,但不会影响其配体亲和力。在重组CHO细胞中,任何两三个N端糖基化位点(Asn63、Asn82、Asn115)的突变都会阻止受体穿梭于质膜[12]。在HEK-293细胞中,GLP-1R胞外域(ECD)存在的信号肽(23个氨基酸)会在GLP-1受体正确加工和运输过程中被切断,以便在质膜形成成熟和完整的糖基化。此外,抑制 hGLP-1R糖基化会阻碍该受体细胞穿膜表达[13,14]。可见,糖基化和信号肽对GLP-1R的穿梭和加工十分重要。
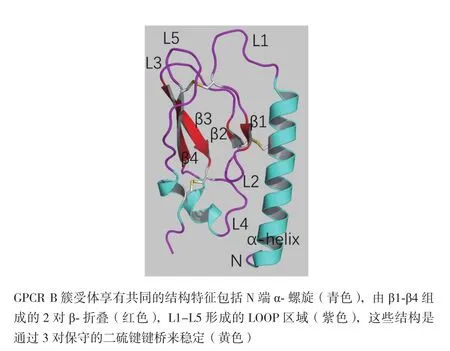
图1 GPCR B簇受体胞外域(ECD)通用结构示意图
1.2 GLP-1R胞外域(ECD)特征
鉴于人们研究GLP-1R全长的长期性和复杂性,单独分离出GLP-1R胞外域(ECD)已经成为了探究GLP-1受体结构和功能的重要手段。1996年,Wilmen等[15]运用大肠杆菌表达ECD(残基20-144)并通过六聚组氨酸标签纯化到可溶性片段。尽管纯化的具有活性功能蛋白也能像GLP-1R全长受体一样同125I-GLP-1(用碘125标记的GLP-1)发生互交作用,但是这些纯化的蛋白处于较低的表达水平,之后用β-巯基乙醇处理发现125I-GLP-1结合受体的能力丧失。2002年,Bazarsuren等[16]也通过大肠杆菌表达出GLP-1R胞外域。与前者不同之处在于表达的蛋白是以包涵体形式存在,后来经过变性、重折叠和复性成功的做出了具有生物活性的GLP-1R胞外域(ECD),实现了蛋白可逆性复性。之后用胃蛋白酶处理发现存在有二硫键,进一步分析证实二硫键的位置分别是Cys46和Cys71、Cys62和Cys104以及Cys85和Cys126。这些具有保守性二硫键对蛋白结构的稳定具有至关重要的意义。2008年,Runge等[17]解析出GLP-1R胞外域同Exendin-4(9-39)形成的复合体,其分辨率为2.2 Å晶体结构。该结构通过N端的α螺旋和两个反向β延伸形成的loop结构阐释了其疏水结合位点并发现了Asp67、Trp72、Pro86、Arg102、Gly108和Trp110六个保守的氨基酸残基。其中Asp67形成分子内相互作用,通过水分子与Arg102相互作用并直接作用于Trp72和Arg121(图2,参照PDB ID:3C5T数据[17])。另外,Asp67还会与Tyr69和Ala70作用来稳定β1和β2链的反转。可见,这些残基对胞外域折叠和受体结构稳定必不可少。2009年,Chritina等[18]解析出GLP-1胞外域同GLP-1形成的复合体,其分辨率为2.2 Å晶体结构。该结构表明激动剂和拮抗剂与受体结合是通过疏水性相互作用来完成的并且这种作用呈现保守性,但是配体结合位点的某些残基赋予了GLP-1的特殊构象(图3-A,3-B,参照PDB ID:3IOL数据[18])。当GLP-1结合受体胞外域时,会呈现出扭曲构象,但从Thr13到Val33展现出连续的α-螺旋结构,这与之前解析的Exendin-4(9-39)与GLP-1R-ECD形成的晶体复合体结构有所不同(图3-C)。2011年,Day等[19]发现GLP-1受体N端域Glu68残基的改变会使GLP-1的亲和力减少8倍,加强GLP-1多肽的C端的正电荷有利于多肽倾向于受体68位氨基酸残基处靠拢,说明Glu68氨基酸对于配体的行为十分重要。最近研究表明,GLP-1受体N端域较大的疏水性作用能够驱使多肽的结合并定位于配体N端激活受体[20]。高蔚丰等[21]通过S52R突变会引起GLP-1受体N端片段活性失活,但缺失前面20个氨基酸以及后面10个氨基酸都不会影响N端域的生物活性,表明Ser52对于维持配体活性至关重要。尽管在探究胞外域结构方面战果累累,然而与所预期的GLP-1R胞外域结构相比,ECD上的很多残基未曾出现过共价修饰。
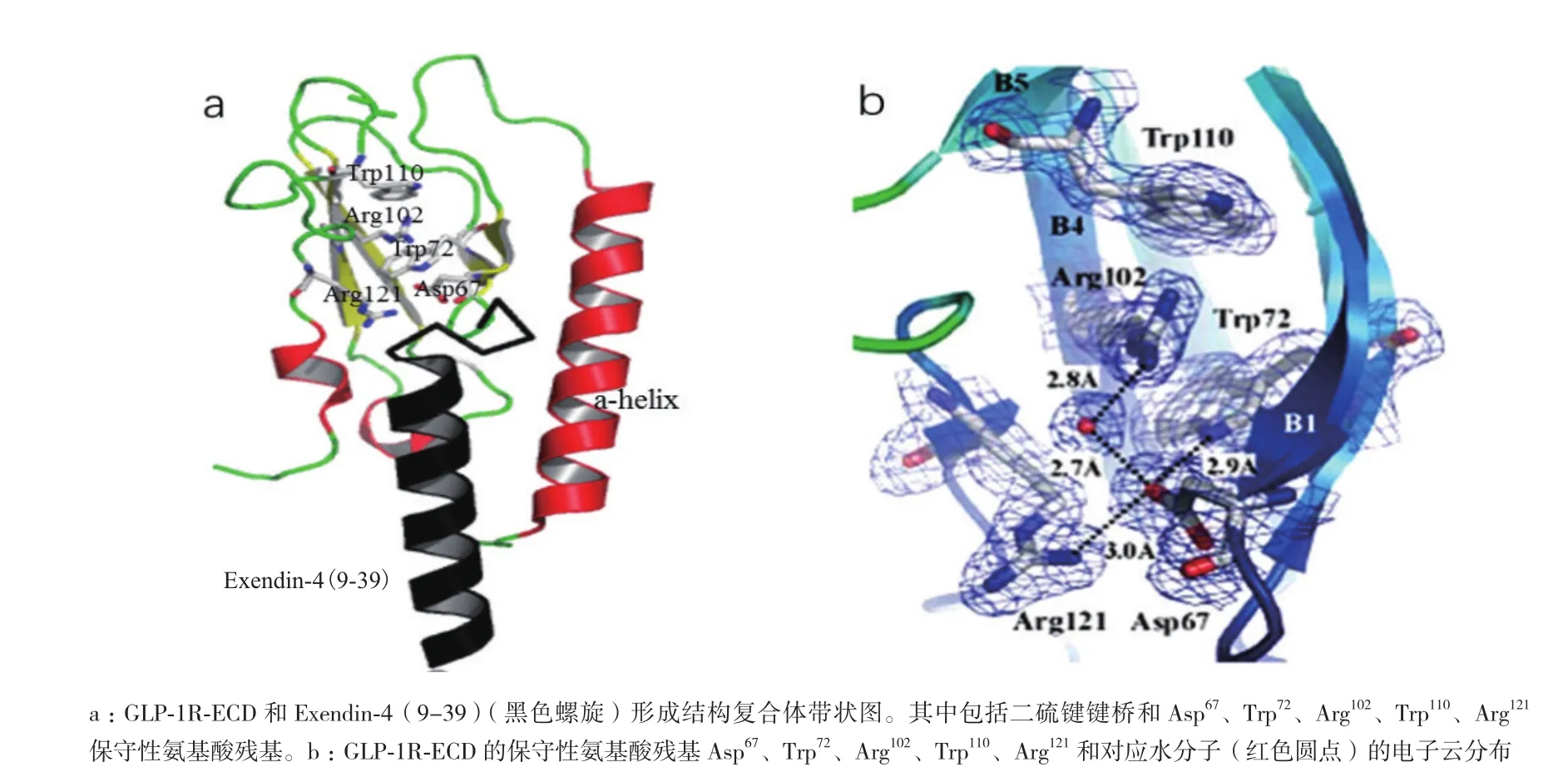
图2 GLP-1R胞外域(ECD)与Exendin-4(9-39)形成复合物结构[17]
1.3 GLP-1R核心域特征
由于受体跨膜域结构的特殊性,人们主要是通过定点诱变以及蛋白互交实验等相关手段来探究其结构。Xiao等[22]对跨膜域 TM2-TM3的5个残基(K197A、D198A、K202A、D215A、R227A) 进 行定点诱变,发现配体结合力存在明显的下降(IC50值相对于WT高出4-20倍)。研究发现,GLP-1受体第二个跨膜螺旋(TMH2)的膜外端的一个残基Asp198[23]和第4个跨膜螺旋(TMH4)的残基Lys288[24]特异性的参与了GLP-1R N端域的结合。将M204A和Y205A进行双突变[25]发现该突变受体同GLP-1的亲和力相对于野生型(WT)受体明显减少了30倍,而Exendin-4只减少了3倍并且对拮抗剂却没有影响,原因可能是这两个位点侧链的疏水性发生了改变。研究调查表明,Ⅱ型糖尿病患者胰岛β细胞中GLP-1受体出现 T149M天然突变会引起GLP-1 结合力下降并且能够降低多肽诱使cAMP调控信号的效能,但cAMP的含量不变。说明这种受体的多态性会影响其结合激动剂的效能[26]。Mann等[27]发现Cys226(TM3)和Cys296(ECL2)之间会形成二硫键,并靠近受体激活“pocket”。进一步通过ECL2上残基定点诱变得出该loop对于激动剂激活受体十分重要。Coopman等[28]发现跨膜域的一些残基(K197A、W284A、R310A等)的突变更倾向与受体效能的降低。人们用点突变和嵌合GLP1R/ GIPR同嵌合GLP-1/GIP 相互作用的分子模拟[29]表明GLP-1的His1和Thr7会同GLP-1受体中的残基Asn302(ECL2)、Ile196(TMH2)和Leu232/Met233(ECL1)相互作用,这为配体与受体结合以及受体激活提供了很重要的线索。Cassandra等[30]通过将跨膜段第2个loop域的相应残基(C296A、D297A、R299A、N300A、N302A、N304A、Y305A和L307A等)进行突变都会影响到GLP-1的结合力和耦合效率及CAMP形成,钙离子内流和胞外信号调控激酶1和2的磷酸化(PERK1/2)的激活,说明第2个loop域的结构对配体结合十分重要,尤其是Trp306Ala会使受体的生物活性丧失。Jin等[31]通过GLP-1R突变发现ECL3上保守残基Arg380与疏水性残基Leu379和Phe381相连可能会同GLP-1的Asp9和Gly4相互作用。后来又用VPAC1R的ECL3去取代GLP-1R的ECL3区域形成GLP-1Rs嵌合受体,结果表明此嵌合受体同GLP-1的结合比较弱,说明GLP-1R受体的ECL3对于受体激活显得十分重要。进一步进行氨基酸点突变表明,GLP-1R受体保守残基Arg380以及疏水性残基Leu379和Phe381可以调控GLP-1的Gly4和Asp9的相互作用。从而提出了配体结合“pocket”主要由GLP-1受体TMH2,ECL1,ECL2和 ECL3的保守性残基构成。近些年,发现了C端对GLP-1R跨膜域表达和内源化产生很大影响,研究表明氨基酸残基411-418对于跨膜域定位于质膜很关键,氨基酸残基419-430可能对于受体耦合Gαs,cAMP诱使产生受体活性十分重要,氨基酸残基431-450对于激动剂诱导hGLP-1R内源化是必不可少的[32]。通过分子模拟发现,受体胞外域靠近跨膜核心区的ECL3并且GLP-1R同配体结合也存在着“closed state”和“open state”两种形式[33],这与之前的胰高血糖素受体(GCGR)与配体结合状况相似[34]。
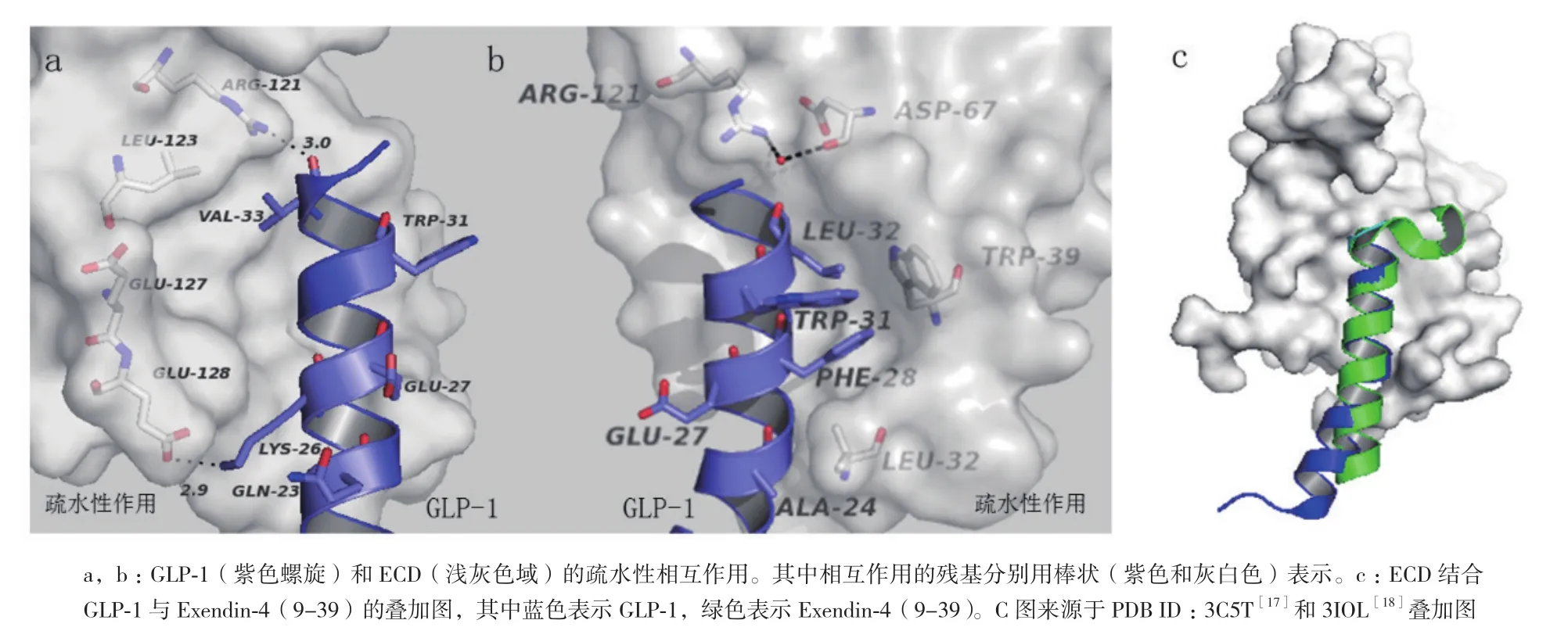
图3 GLP-1R胞外域(ECD)同GLP-1疏水性相互作用
2 GLP-1R的功能相关性研究
2.1 受体激活
GLP-1R属于G蛋白偶联受体B簇亚类。GPCR B簇受体与配体作用机制主要是被大家公认的“twodomain model”[35](图4)。配体的C端螺旋首先同GLP-1R胞外域(ECD)结合,从而确保配体的N端同GLP-1受体的核心区域(TMD)进行二次交合,后者相互作用对于激动剂激活受体至关重要[35]。因此提出了解释GPCR B簇受体激活模型,即配体结合诱使内源性激动剂构象改变,然后同受体核心域相互作用产生活性[36]。已经报道了配体的α螺旋二级结构在二域模型中调控受体胞外域ECD的起始作用十分重要,并提出了螺旋包被模型来解释GPCR B簇不同配体是怎样激活受体[9]。
GLP-1和Exendin-4都是α-螺旋的多肽,能与GLP-1R胞外域多个接触位点相互作用诱导受体信号。它们的两性特征决定了它们同受体ECD相互作用呈现出保守性,其中配体的疏水面是相互作用的关键并且受体激动剂多肽疏水区形成的微小结合能对结合最为关键。受体激活模型的第二阶段表明,ECD对接多肽直接促进配体N端同受体核心“pocket”相互作用引起跨膜α-螺旋构象重排,诱使膜内Loop环刺激胞内信号传导。

图4 G蛋白偶联受体与其配体结合的“two-domain model”
2.2 GLP-1R信号传导调控机制
一般来说,GLP-1受体N端主要是识别特异性的配体,但是受体的核心区对于信号特异性传导发挥重要作用。GLP-1R属于G蛋白偶联受体中Gs亚类。它是一种多效性偶联受体,主要通过与多种G蛋白(Gαs、Gαi、Gαo和Gαq/11)偶联来调控细胞通路。当与GLP-1结合后,G蛋白α亚基与β、γ亚基解离并对不同信号通路进行介导[7]。在β细胞中偶联Gαs蛋白,激活腺苷环化酶,促使cAMP在细胞内含量升高并增加蛋白激酶A(PKA)和cAMP-2激活交换蛋白(Epac2)的含量,引起离子通道活性改变,钾离子通道关闭,电压依赖性钙离子通道打开(VDCCs),钙离子内流,胰岛素原基因转录增加,胰岛素分泌小泡释放。GLP-1可以通过cAMPPKA途径提高葡萄糖感受性,刺激血糖依赖性胰岛素的分泌[37]。除此之外,GLP-1R受体还可以通过G蛋白β、γ亚基来激活磷脂酰基醇-3-激酶(PI3K)和丝裂原活化蛋白激酶(MAPK)的信号通络诱导β细胞的增值和分化;另外,cAMP还可以以不依赖PKA的方式,通过与β细胞中cAMP调节的鸟苷酸交换因子(cAMP-regulated guaninenucleotide exchange factors,cAMP-GEFs)相互作用,激活Ras/MAPK[38](mitogen-activated protein kinase)信号通路,促进β细胞的生长和分化[38]。不仅如此,GLP-1R还能够通过调控cAMP反应元件结合蛋白(CREB)和蛋白复活因子Bcl-2、Bcl-XL来抑制细胞凋亡(图5)。近些年也发现β-arrestin的募集也参与了GLP-1R功能[39]。如敲出β细胞中β-arrestin1会诱使cAMP的降低和胰岛素释放减少80%[40],敲出β-arrestin2的野生小鼠会产生餐后高血糖,糖耐性降低并会引起胰岛素抵抗等症状[41]。研究表明[42,43],β-arrestins主要是通过形成β-arrestins依赖性信号复合物和信号分子(ERK 1/2,JNK3)作为信号载体传递信号于MAPK,从而调控β-arrestins依赖性胰岛素信号通路。

图5 胰岛β细胞中GLP-1R调控的信号通路
2.3 GLP-1R生理功能
GLP-1R作为糖尿病治疗的重要靶点。在机体内广泛表达由于分布于胃、小肠、心脏、肾脏、肺及大脑等组织。在胰岛细胞中,GLP-1R主要是促进胰岛素的释放,增加胰岛β细胞的再生,抑制β细胞的凋亡,降低胰高血糖素的释放。在胃肠道等组织中[44],GLP-1R可以通过与其激动剂结合抑制胃肠道的蠕动和胃液分泌,延迟胃的排空,增加饱食感。在神经组织中,小分子GLP-1R激动剂能够穿透于大脑激活GLP-1R表达的神经元子集,保护神经细胞的凋亡和加强学习记忆能力[45]。不仅如此,GLP-1R也能够控制食物摄取来减轻体重[46]。在心血管方面,通过对2个月的鼠进行GLP-1R基因的敲出发现其静息心率降低,左心室舒张压升高[47];除此之外,GLP-1R还能调控降低氧化应激压和抑制心肌细胞的凋亡[48]。
3 小分子药物的筛选
GLP-1R非肽激动剂因具有潜在口服活性能避免T2DM患者长期自我注射,因而引起了人们的广泛关注。目前,已报道的有关基于GLP-1受体治疗糖尿病药物而开发的小分子先导化合物较多(表1)。
3.1 小分子拮抗剂
事实上,第一个GLP-1R非肽类配体T-0632实际上是一种拮抗剂[49]。相比较125I-Exendin-4(9-39)而言,它同hGLP-1R的IC50为1.2 μmol/L并且在COS-7细胞中能够抗拒GLP-1诱导产生cAMP。W33S突变会使得这种拮抗剂同受体结合力降低100倍,表明它同ECD的结合位点为Trp33,该氨基酸位于GLP-1受体胞外域α-螺旋中但不参与多肽的结合。尽管这种化合物能够作为很好的分子工具被用来研究机体中的生理化学特征,但是结合力相对较弱以致不足以用来研究GLP-1受体。Catein是一种天然的多酚类物质[50],能够作为GLP-1R选择性的负调控变构调节剂。在钙离子流动实验中,Catein不会对激动剂调控信号产生影响。但是在cAMP积累实验中,Catein却发挥着GLP-1(7-36)NH2和GLP-1(1-36)NH2的负调控调节剂的作用。例如,Catein会降低由cAMP引起的GLP-1信号传导效能,但对由GLP-1受体多肽(Exendin-4)引起的非cAMP信号通路不起调控作用[50]。
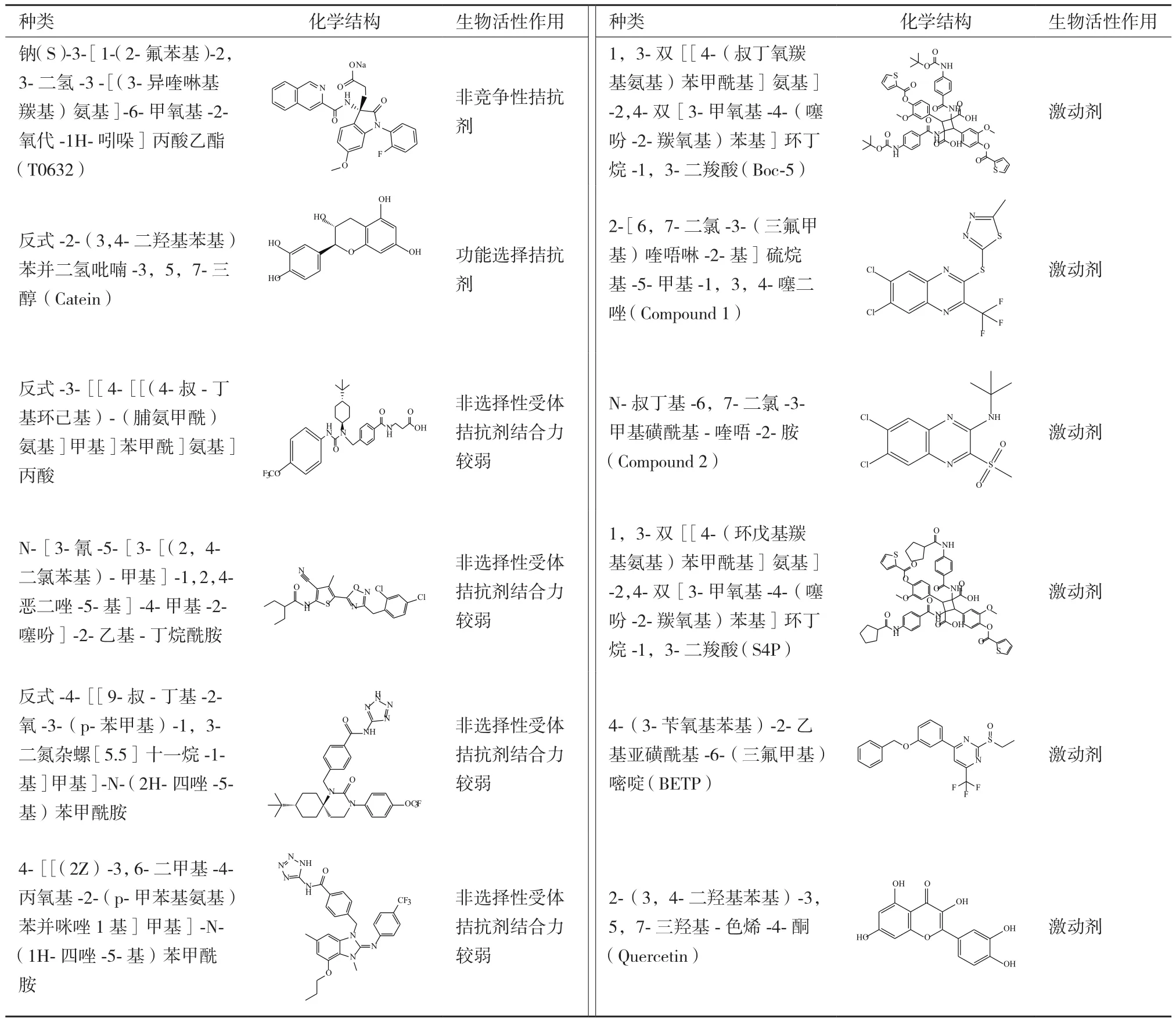
表1 基于GLP-1受体的先导化合物类别[58]
3.2 小分子激动剂
2007年,人们通过高通量筛选方法从多达48 000个的合成和天然化合物中筛选出两个GLP-1R非肽激动剂,Boc5[51]和它的类似物S4P。它们会在DMSO长期储存液中自发形成二聚化合物。基于CRE-荧光素酶分析表明,Boc5在HEK-293细胞表达rGLP-1R中扮演着完全激动剂的角色并具有1.08 μmol/L的效能,但是与其结构相似的小分子S4P却是一种部分激动剂。然而,在cAMP积累实验中却发现Boc5和S4P都是部分激动剂。更加重要的是,用Boc5和S4P代替125I-GLP-1进行受体结合实验也会引起Exendin-4(9-39)功能性拮抗作用。进一步实验表明,Boc5能够促进分离的鼠胰岛糖依赖性胰岛素的分泌,抑制鼠食物摄取并能够减少db/db小鼠HbA1c含量到正常水平。由于它们缺乏类药分子所具有的结构特征(背离了Lipinsky和Veber规律),Boc5只是作为一种有用的概念性分子,并且有关该分子结合受体的机理可能同GLP-1类似[52]。2007年发现另外一种非肽类激动剂,compound2(Cmp2)及其类似结构物[53]。这些化合物较Boc5分子量小并且具有不同的药理学特性。它们是GLP-1R的一种变构调节剂,因为它们不仅能够独立发挥调节作用而且也能够作为变构调节剂加强天然激动剂的活性。然而,这种变构调节剂只是局限于增加GLP-1亲和力(26倍)而对其效能却无任何作用。但是,在hGLP-1R表达的FIpIn-CHO细胞中有助于提高GLP-1的效能[54]。Cmp2还能够刺激正常小鼠胰岛糖依赖性胰岛素的释放但是对于GLP-1R敲出的小鼠却效用较小。Cmp2和GLP-1对GLP-1R表达的HEK-293细胞产生影响表明这两种激动剂都是通过Gαs发挥调控作用[55]。进一步研究表明EX4(9-39)会抑制Cmp2调控受体内源化。最新研究表明Cmp2结合GLP-1R会引起受体偶联Gαs构象改变,但不会引起钙离子聚集,ERK磷酸化以及受体内源化[56]。黄酮类化合物是另外一种GLP-1R的激动剂。尽管它不会直接激活GLP-1R受体,但可以通过GLP-1和Exendin-4加强钙离子调控反应。黄铜类结构表明3-OH基团是发挥活性的关键基序[57]。
4 展望
已知有大约80%多肽类激素都是依靠激活GPCR信号通路来调控生理机能。因此,GPCR紊乱会引起人类的一些疾病。目前,有大约50%的药物靶点都属于GPCR[59]。GPCR B簇成员GLP-1R作为治疗2-型糖尿病的重要靶点,对于该受体与配体的相互作用的研究显得尤为重要。但是,有关配体结合受体核心区的位点目前还是停留在推测阶段,受体激活的分子机制尚不明朗。但是有关GPCR B簇受体GCGR,CRF1R[60,61]相继被解析,这为GLP-1R分子结构作用机理研究提供了较好的借鉴。尽管有关基于GLP-1受体药物发展很多,但是还不曾有能够有效口服GLP-1R小分子受体激活剂被用于治疗。让人更加欣慰的是,目前有关小分子GLP-1受体配基的鉴定步伐正在加速。尽管多肽结合机理和GLP-1R受体激活很复杂以致于模拟小分子化合物十分困难,但是通过变构模型促使小分子发挥效用的实例[52]表明针对靶点的药物化学策略是可行的。因此,运用先进的结构生物方法学和更复杂分析系统及测试方案包括理解受体配体偏向性信号传导将有可能推进类药分子的发展。这也有助于为下一代糖尿病药物研发利用提供参考,具有十分重要的临床应用价值。
[1] Blad CC, Tang C, Offermanns S. G protein-coupled receptors for energy metabolites as new therapeutic targets[J]. Nature Reviews Drug Discovery, 2012, 11:603-619.
[2] 田洪斌. 注射用艾塞那肽冻干粉针剂的研究[D]. 长春:吉林大学, 2008.
[3] Koole C, Savage EE, Christopoulos A, et al. Minireview:Signal bias, allosterism, and polymorphic variation at the GLP-1R:implications for drug discovery[J]. Molecular Endocrinology, 2013, 27:1234-1244.
[4] Lauritsen KB, Moody AJ, Christensen KC, et al. Gastric inhibitory polypeptide(GIP)and insulin release after small-bowel resection in man[J]. Scandinavian Journal of Gastroenterology, 1980, 15:833-840.
[5] Thorens B, Widmann C. Structure and function of the glucagon-like peptide-1 receptor[M]. Springer Berlin Heidelberg, 1996:255-273.
[6] Brubaker PL, Drucker DJ. Structure-function of the glucagon receptor family of G protein-coupled receptors:the glucagon, GIP, GLP-1, and GLP-2 receptors[J]. Receptors & Channels, 2011, 8:179-188.
[7] Sloop FSW, Kyle W. Physiology and emerging biochemistry of the glucagon-like peptide-1 receptor[J]. Experimental Diabetes Research, 2012, 2012:344-350.
[8] Pabreja K, Mohd MA, Koole C, et al. Molecular mechanisms underlying physiological and receptor pleiotropic effects mediated by GLP-1R activation[J]. British Journal of Pharmacology, 2014, 171:1114-1128.
[9] Parthier C, Reedtz-Runge S, Rudolph R, et al. Passing the baton in class B GPCRs:peptide hormone activation via helix induction?[J]. Trends in Biochemical Sciences, 2009, 34:303-310.
[10] Parthier C, Kleinschmidt M, Neumann P, et al. Crystal structure of the incretin-bound extracellular domain of a G protein-coupled receptor[J]. Proceedings of the National Academy of Sciences of the United States of America, 2007, 104:13942-13947.
[11] Göke R, Just R, Lankat-Buttgereit B, et al. Glycosylation of the GLP-1 receptor is a prerequisite for regular receptor function[J]. Peptides, 1994, 15:675-681.
[12] Chen Q, Miller LJ, Dong M. Role of N-linked glycosylation in biosynthesis, trafficking, and function of the human glucagon-like peptide 1 receptor[J]. Ajp Endocrinology & Metabolism, 2010, 299:E62-68.
[13] Aiysha T, Venkateswarlu K. The regions within the N-terminus critical for human glucagon like peptide-1 receptor(hGLP-1R)cell surface expression[J]. Scientific Reports, 2014, 4:7410.
[14] Huang Y, Wilkinson GF, Willars GB. Role of the signal peptide in the synthesis and processing of the glucagon-like peptide-1 receptor[J]. Experimental Biology & Medicine, 2007, 67:141-145.
[15] Wilmen A, Goke B, Göke R. The isolated N-terminal extracellular domain of the glucagon-like peptide-1(GLP)-1 receptor has intrinsic binding activity[J]. Febs Letters, 1996, 398(45):43-47.
[16] Bazarsuren AGU, Wozny M, Reusch D, et al. In vitro folding, functional characterization, and disulfide pattern of the extracellular domain of human GLP-1 receptor[J]. Biophysical Chemistry, 2002, 96:305-318.
[17] Steffen R, Henning TG, Kjeld M, et al. Crystal structure of the ligand-bound glucagon-like peptide-1 receptor extracellular domain[J]. Journal of Biological Chemistry, 2008, 283:11340-11347.
[18] Christina Rye U, Patrick G, Lotte Bjerre K, et al. Crystal structure of glucagon-like peptide-1 in complex with the extracellular domain of the glucagon-like peptide-1 receptor[J]. Journal of Biological Chemistry, 2010, 285:723-730.
[19] Day JW, Li P, Patterson JT, et al. Charge inversion at position 68 of the glucagon and glucagon-like peptide-1 receptors supports selectivity in hormone action[J]. Journal of Peptide Science, 2011, 17:218-225.
[20] Patterson JT, Li P, Day JW, et al. A hydrophobic site on the GLP-1 receptor extracellular domain orients the peptide ligand for signal transduction[J]. Molecular Metabolism, 2013, 2:86-91.
[21] 高蔚丰, 王娟. 保守的第52位色氨酸突变引起的胰高血糖素样肽1受体N端片段活性丧失[J]. 生物工程学报, 2013, 29:87-94.
[22] Xiao Q, Jeng W, Wheeler MB. Characterization of glucagonlike peptide-1 receptor-binding determinants[J]. Journal of Molecular Endocrinology, 2000, 25:321-335.
[23] Maturana RLD, Dan D. The glucagon-like peptide-1 receptor binding site for the N-terminus of GLP-1 requires polarity at Asp198 rather than negative charge[J]. Febs Letters, 2002, 530:244-248.
[24] Al-Sabah S, Donnelly D. The positive charge at Lys-288 of the glucagon-like peptide-1(GLP-1)receptor is important for binding the N-terminus of peptide agonists-FEBS Letters[J]. Febs Letters, 2003, 553:342-346.
[25] Rakel LDM, Janet TB, Fatima A, et al. Met-204 and Tyr-205 are together important for binding GLP-1 receptor agonists but not their N-terminally truncated analogues[J]. Protein & Peptide Letters, 2004, 11:15-22.
[26] Beinborn M, Worrall CI, Mcbride EW, et al. A human glucagonlike peptide-1 receptor polymorphism results in reduced agonist responsiveness[J]. Regulatory Peptides, 2005, 130:1-6.
[27] Mann RJ, Suleiman AS, Rakel López DM, et al. Functional coupling of Cys-226 and Cys-296 in the glucagon-like peptide-1(GLP-1)receptor indicates a disulfide bond that is close to the activation pocket[J]. Peptides, 2010, 31:2289-2293.
[28] Coopman K, Wallis R, Robb G, et al. Residues within the transmembrane domain of the glucagon-like peptide-1 receptor involved in ligand binding and receptor activation:modelling the ligand-bound receptor[J]. Molecular Endocrinology, 2011, 25:1804-1818.
[29] Jin MM, Hee Young K, Sumi P, et al. Evolutionarily conserved residues at glucagon-like peptide-1(GLP-1)receptor core confer ligand-induced receptor activation[J]. Journal of Biological Chemistry, 2011, 287:3873-3884.
[30] Cassandra K, Denise W, John S, et al. Second extracellular loop of human glucagon-like peptide-1 receptor(GLP-1R)has a critical role in GLP-1 peptide binding and receptor activation[J]. Journal of Biological Chemistry, 2012, 287:3642-3658.
[31] Jin MM, Yoo-Na L, Sumi P, et al. Ligand binding pocket formed by evolutionarily conserved residues in the glucagon-like peptide-1(GLP-1)receptor core domain[J]. Journal of Biological Chemistry, 2015, 290:5696-5706.
[32] Thompson A, Kanamarlapudi V. Distinct regions in the C-Terminus required for GLP-1R cell surface expression, activity and internalisation[J]. Molecular & Cellular Endocrinology, 2015, 413:66-77.
[33] Zhang J, Gu S, Sun X, et al. Computational insight into conformational states of glucagon-like peptide-1 receptor(GLP-1R)and its binding mode with GLP-1[J]. Rsc Advances, 2016, 6:13490-13497.
[34] Yang L, Yang D, De GC, et al. Conformational states of the fulllength glucagon receptor[J]. Nature Communications, 2015, 6:2708-2713.
[35] Hoare SRJ. Mechanisms of peptide and nonpeptide ligand binding to Class B G-protein-coupled receptors[J]. Drug Discovery Today, 2005, 10:417-427.
[36] Dong M, Gao F, Pinon DI, et al. Insights into the structural basis of endogenous agonist activation of family B G protein-coupled receptors[J]. Molecular Endocrinology, 2008, 22:1489-1499.
[37] Holz GG, Leech CA, Heller RS, et al. cAMP-dependent mobilization of intracellular Ca2+stores by activation of ryanodine receptors in pancreatic beta-cells. A Ca2+signaling system stimulated by the insulinotropic hormone glucagon-like peptide-1-(7-37)[J]. Journal of Biological Chemistry, 1999, 274:14147-14156.
[38] Leech CA, Holz GG, Chepurny O, et al. Expression of cAMP-regulated guanine nucleotide exchange factors in pancreatic β-cells[J]. Biochemical & Biophysical Research Communications, 2000, 278:44-47.
[39] Noriyuki S, Takeshi I, Takeshi Y, et al. Beta-Arrestin-1 mediates glucagon-like peptide-1 signaling to insulin secretion in cultured pancreatic beta cells[J]. Proceedings of the National Academy of Sciences of the United States of America, 2008, 105:6614-6619.
[40] Shukla AK, Xiao K, Lefkowitz RJ. Emerging paradigms of β-arrestin-dependent seven transmembrane receptor signaling[J]. Trends in Biochemical Sciences, 2011, 36:457-469.
[41] Luan B, Zhao J, Wu H, et al. Deficiency of a β-arrestin2 signal complex contributes to insulin resistance[J]. Nature, 2009, 457:1146-1149.
[42] Dalle S, Ravier MA, Bertrand G. Emerging roles for β-arrestin-1 in the control of the pancreatic β-cell function and mass:New therapeutic strategies and consequences for drug screening[J]. Cellular Signalling, 2011, 23:522-528.
[43] Etienne K, Ljiljana N, May S, et al. Differential regulation of endosomal GPCR/β-arrestin complexes and trafficking by MAPK[J]. Journal of Biological Chemistry, 2014, 289:23302-23317.
[44] Koliaki C, Doupis J. Incretin-based therapy:a powerful and promising weapon in the treatment of type 2 diabetes mellitus[J]. Diabetes Therapy, 2011, 2:101-121.
[45] During MJ, Cao L, Zuzga DS, et al. Glucagon-like peptide-1 receptor is involved in learning and neuroprotection[J]. Nature Medicine, 2003, 9:1173-1179.
[46] Baggio LL, Drucker DJ. Glucagon-like peptide-1 receptors in the brain:controlling food intake and body weight[J]. Journal of Clinical Investigation, 2014, 124:4223-4226.
[47] Robert G, Xiaomang Y, Baggio LL, et al. Cardiac function in mice lacking the glucagon-like peptide-1 receptor[J]. Endocrinology, 2003, 144:2242-2252.
[48] Laviola L, Leonardini A, Melchiorre M, et al. Glucagon-like peptide-1 counteracts oxidative stress-dependent apoptosis of human cardiac progenitor cells by inhibiting the activation of the c-Jun N-terminal protein kinase signaling pathway[J]. Endocrinology, 2012, 153:5770-5781.
[49] Tibaduiza EC, Chen C, Beinborn M. A small molecule ligand of the glucagon-like peptide 1 receptor targets its amino-terminal hormone binding domain[J]. Journal of Biological Chemistry, 2001, 276:37787-37793.
[50] Denise W, John S, Cassandra K, et al. Modulation of the glucagonlike peptide-1 receptor signaling by naturally occurring and synthetic flavonoids[J]. Journal of Pharmacology & Experimental Therapeutics, 2011, 336:540-550.
[51] Chen D, Liao J, Li N, et al. A nonpeptidic agonist of glucagon-like peptide 1 receptors with efficacy in diabetic db/db mice[J]. Proceedings of the National Academy of Sciences of the United States of America, 2007, 104:943-948.
[52] Lin F, Wang R. Molecular modeling of the three-dimensional structure of GLP-1R and its interactions with several agonists[J]. Journal of Molecular Modeling, 2008, 15:53-65.
[53] Knudsen LB, Kiel D, Teng M, et al. Small-molecule agonists for the glucagon-like peptide 1 receptor[J]. Proceedings of the National Academy of Sciences of the United States of America, 2007, 104:937-942.
[54] Koole C, Wootten D, Simms J, et al. Allosteric ligands of the glucagon-like peptide 1 receptor(GLP-1R)differentially modulate endogenous and exogenous peptide responses in a pathway-selective manner:implications for drug screening[J]. Molecular Pharmacology, 2010, 78:456-465.
[55] Coopman K, Huang Y, Johnston N, et al. Comparative effects of the endogenous agonist Glucagon-Like Peptide-1(GLP-1)-(7-36)amide and the small-molecule ago-allosteric agent "compound 2" at the GLP-1 receptor[J]. Journal of Pharmacology & Experimental Therapeutics, 2010, 334:795-808.
[56] Thompson A, Stephens JW, Bain SC, et al. Molecular characterisation of small molecule agonists effect on the human glucagon like peptide-1 receptor internalisation[J]. PLoS One, 2016, 11:e0154229.
[57] Wootten D, Simms J, Koole C, et al. Modulation of the glucagon-like peptide-1 receptor signaling by naturally occurring and synthetic flavonoids[J]. Journal of Pharmacology & Experimental Therapeutics, 2011, 336:540-550.
[58] Willard FS, Bueno AB, Sloop KW. Small molecule drug discovery at the glucagon-like peptide-1 receptor[J]. Journal of Diabetes Research, 2012, 2012:344-350.
[59] Millar RP, Newton CL. The year in G protein-coupled receptor research[J]. Molecular Endocrinology, 2009, 24:261-274.
[60] Siu FY, He M, de Graaf C, et al. Structure of the human glucagon class B G-protein-coupled receptor[J]. Nature, 2013, 499:444-449.
[61] Hollenstein K, Kean J, Bortolato A, et al. Structure of class B GPCR corticotropin-releasing factor receptor 1[J]. Nature, 2013, 499:438-443.
(责任编辑 马鑫)
Research Progress on Structure and Function of GLP-1R and Screening for Small Molecule Drugs
HU Zhong-ping CHENG Nian YANG Fan SU Zheng-ding
(Institute of Biomedical and Pharmaceutical Sciences,Hubei Collaborative Innovation Center for Industrial Fermentation,the Key Laboratory of Industrial Fermentation of Ministry of Education,Hubei University of Technology,Wuhan 430068)
Glucagon-like peptide-1 receptor(GLP-1R)as an important target for type 2 Diabetes mellitus(T2DM)therapy,presents clinic significance. The breakthroughs on GLP-1R structures and functions have been made via structural biology and protein engineering. However,it is still unknown on the analysis of its full length structure,the molecular mechanism of polypeptide binding receptors,and the intrinsic mechanism of receptor activation. Owing to the rapid research progresses relating to GLP-1R,this article briefly describes the structure and function of the GLP-1 receptor and the leading compound of existing small molecule drugs,also discusses the developing direction and application prospects of action mechanism of the GLP-1 receptor molecule structure,aiming to provide structure base for the treatment of T2DM.
GLP-1R;molecular structure;small molecule drugs
10.13560/j.cnki.biotech.bull.1985.2017.02.005
2016-05-23
武汉市自然科学基金重点项目(2015060101010033)
胡中平,男,硕士研究生,研究方向:蛋白质结构;E-mail:15623601030@163.com
苏正定,男,教授,研究方向:蛋白质工程与生物医药;E-mail:zhengdingsu@mail.hbut.edu.cn
