Quality assessment of Chrysanthemum indicum Flower by simultaneous quantifcation of six major ingredients using a single reference standard combined with HPLC fngerprint analysis
2017-01-19JiaoHeXiaoxueWuYaliKuangTianyangWangKaishunBiQingLi
Jiao He,Xiaoxue Wu,Yali Kuang,Tianyang Wang, Kaishun Bi,Qing Li,*
aSchool of Pharmacy,Shenyang Pharmaceutical University,Shenyang 110016,China
bNational and Local United Engineering Laboratory for Key Technology of Chinese Material Medical Quality Control,Shenyang Pharmaceutical University,Shenyang 110016,China
Quality assessment of Chrysanthemum indicum Flower by simultaneous quantifcation of six major ingredients using a single reference standard combined with HPLC fngerprint analysis
Jiao Hea,b,Xiaoxue Wua,b,Yali Kuanga,b,Tianyang Wanga,b, Kaishun Bia,b,Qing Lia,b,*
aSchool of Pharmacy,Shenyang Pharmaceutical University,Shenyang 110016,China
bNational and Local United Engineering Laboratory for Key Technology of Chinese Material Medical Quality Control,Shenyang Pharmaceutical University,Shenyang 110016,China
A R T I C L EI N F O
Article history:
Received 28 May 2015
Received in revised form 14 August 2015
Accepted 24 August 2015
Available online 10 September 2015
Chrysanthemum indicumFlower
Single standard to determinate
multi-components
Fingerprint
Quality assessment
Chrysanthemum indicumFlower is usually consumed as functional food.This paper described an improved total quality assessment method forChrysanthemum indicumFlower by simultaneous quantitation using a single standard to determine multi-components method combined with high performance liquid chromatography fngerprint analysis.Six main components ofChrysanthemum indicumFlower including two favonoids and four phenolic acids were simultaneously quantifed using linarin as the internal reference standard.The method was fully validated with respect to linearity,precision,accuracy,robustness and stability. The validated method was successfully applied to the analysis of thirty three batches of
Chrysanthemum indicumFlower samples.Under the same chromatographic conditions,fngerprint analysis in combination with Similarity analysis and principal component analysis was performed to identify the samples from different regions.In general,an effective assessment using a single standard to determinate multi-components method combined with fngerprint analysis make the reliable qualitation and quantitation analysis ofChrysanthemum indicumFlower available.
©2016 The Authors.Production and hosting by Elsevier B.V.on behalf of Shenyang Pharmaceutical University.This is an open access article under the CC BY-NC-ND license
(http://creativecommons.org/licenses/by-nc-nd/4.0/).
1.Introduction
Chrysanthemum indicumFlower(CIF)is a well-known edible and medicinal plant with small yellow fowers.Flowers and buds of CIF are widely used as a food supplement,or herbal tea, which are considered as the health food by many consumers.It is also widely used to treat various immune-related disorders,hypertension symptoms and several infectious diseases such as stomatitis and fever in folk medicine in China and Korea for centuries[1–3].CIF is known to contain several classes of biologically active compounds including essential oils,terpenoids,favonoids,and phenolic acids[4,5].Among the nonvolatile oil,the major active components include favonoids of luteolin-7-O-β-D-glucoside and linarin,as well as phenolic acids of chlorogenic acid,3,5-di-O-caffeoylquinic acid, 3,4-di-O-caffeoylquinic acid and 4,5-di-O-caffeoylquinic acid. The phenolic acids have antibacterial,antiphlogistic,antimutagenic,antioxidant and other biological activities[6].Linarin and luteolin-7-O-β-D-glucoside are used as remedies because of their antiphlogistic,spasmolytic,good antioxidant and free radical scavenging properties[7].
In recent years,there have been more and more applications for CIF extracts especially in the nutraceutical and food areas.Since distributed widely in China,the quality of CIF differs.Therefore,a reasonable assessment method for CIF is urgently required.In the past decades,a huge number of analytical strategies have been designed to assess the quality of natural dietary supplements(NDSs).These involved quantifcation of a single compound or multiple components,as well as fngerprint analysis.Single marker compound quantifcation is simple and convenient,but it does not afford suffcient quantitative information for the other bioactive components in complex NDSs[8].Meanwhile,fngerprint analysis can control the quality consistency and stability of food products[9],but it cannot provide accurate quantifcation of analytes in NDSs [10].Thus,it is reasonable and essential to combine multicomponent determination with fngerprint analysis to control the total quality of NDSs.However,the limited availability of various chemical standard substances for quantitative analysis is a major hurdle[11].On assessing the alternative measures available for the practical comprehensive determination of NDSs,the single standard of determination multiple components(SSDMC)method is worth considering[12,13].Through the above analysis,it is necessary to perform the method based on SSDMC method and fngerprint analysis in routine quality control of CIF.In the process,techniques such as HPLC[14], gas chromatography(GC)[15],high performance capillary electrophoresis(HPCE)[16],gas chromatography–mass spectrometry (GC–MS)[17]and liquid chromatography–mass spectrometry (LC–MS)are often used.However,HPLC is simple,reliable and inexpensive,and has been widely used for quality control of herbal dietary supplements.
In this study,HPLC chromatogram with a good separation and symmetrical peak shape was achieved.The highest content of six components in CIF included chlorogenic acid,luteolin-7-O-β-D-glucoside,3,4-di-O-caffeoylquinic acid,3,5-di-O-caffeoylquinic acid,4,5-di-O-caffeoylquinic acid and linarin,the peak areas of which were above 75%of the total.Thus,the simultaneous quantifcation of the six active compounds would be of signifcant value for the quality control of CIF,but to date, no reports have been published.We present here a simple and reliable HPLC method,which allows the simultaneous quantifcation of the six major components using linarin as the internal reference standard.The new method was fully validated,and the results were compared with those obtained from traditional external standard method.The developed method was successfully applied to the quantitative analysis of 33 batches of CIF from different regions.Meanwhile the HPLC fngerprints of CIF were established under the same chromatographic conditions.Sixteen peaks in the chromatography were marked as the common peaks.Multivariate statistical analysis technique including principal component analysis(PCA)and similarity analysis(SA)were used to differentiate between commercial CIF samples.The present study involves a combination of CIF quantitation of 6 chemical constituents,fngerprints,SA and PCA.This combination offers a method to ensure quality consistency of commercial samples.
2.Materials and methods
2.1.Instrumentation and chromatographic conditions
Shimadzu 20A HPLC System(Shimadzu Corporation,Japan) comprised a binary solvent delivery system,an on-line degasser, an auto-sampler,a column temperature controller and a photodiode array detector coupled with Lab Solution software. Additional different HPLC instrument was used.Agilent 1260 HPLC system compromised a quaternary solvent delivery system,an on-line degasser,an auto-sampler,a column temperature controller and a photodiode array detector coupled with an analytical workstation.KH5200b sonicated bath(He Chuang,Kun Shan,Co.Ltd)was used for sample preparation. A BP 211D balance(Sartorius,Germany)was used to weigh the samples.
The separation was performed on a Luna C18column(4.6 mm×250 mm,5 μm,Phenomenex Inc,CA,USA)protected by a Security Guard C18 guard column(4.0 mm×3.0 mm,5 μm, Phenomenex Inc.)with a sample injection volume of 10 μl.Detection wavelength was set at 326 nm.The fow rate was 0.8 ml/min.The column temperature was maintained at 20oC. The mobile phase consisted of 0.05%phosphoric acid in water (A)and acetonitrile(B).The gradient program was as follows: 12–19%(B)in 0–17 min,19–20%(B)in 17–30 min,20–21%(B)in 30–40 min,21–23%(B)in 40–50 min,23–25%(B)in 50–57 min, 25–26%(B)in 57–70 min.
2.2.Chemicals and standards
Acetonitrile for HPLC was purchased from Fisher Scientifc (USA).Water for HPLC was redistilled.Analytical grade phosphoric acid was used for HPLC.Methanol used for extraction was purchased from Fisher Scientifc(USA).Gallic acid, chlorogenic acid,cryptochlorogenic acid,caffeic acid,3,4-di-O-caffeoylquinic acid,3,5-di-O-caffeoylquinic acid,4,5-di-O-caffeoylquinic acid,luteolin-7-O-β-D-glucoside and linarin were purchased from Chengdu Must Biological Technology Co.Ltd (Chengdu,China).The purities of all the chemical reference substances were more than 98%.
2.3.Materials
Thirty three batches of representative samples of CIF were collected from different regions involving thirteen provinces of China(Table 4),and all were identifed by Professor Jia Ying, who is in the School of Chinese Material Medica at Shenyang Pharmaceutical University.All these samples have been harvested in the fall of 2012 and were obtained from commercial sources and through online.
2.4.Preparation of standard solutions
Stock solutions of the reference standards(Gallic acid, Chlorogenic acid,Cryptochlorogenic acid,caffeic acid,3,4-di-O-caffeoylquinic acid,3,5-di-O-caffeoylquinic acid,4,5-di-O-caffeoylquinic acid,luteolin-7-O-β-D-glucoside and linarin)were prepared by dissolving accurately weighed standards in methanol and a little dimethyl sulfoxide to yield the concentrations of 0.6812,0.7156,0.4516,0.3412,0.7376,0.928,1.170,0.935, 1.211 mg/ml respectively,and stored in a 10 ml volumetric fask. The stock solution was then diluted with methanol to the appropriate concentration range to establish calibration curves.
2.5.Preparation of sample solutions
0.5 g of the powdered CIF was sonicated in 20 ml of 60%methanol for 30 min.The solution was then transferred to a 10 mL volumetric fask.Prior to the sample injection,an adequate volume was passed through a 0.45 μm PTFE membrane flter and the frst portion of the fltrate was discarded.The subsequent fltrate was stored as the sample solution.
2.6.Calculation of relative response factor and relative retention time
The RRF of reference standard X(RRFx)was calculated based on the linearity data.It was briefy described as following two equations:The ABiand Axiare the peak areas of the internal single standard(linarin)and the other reference standards(X), at the concentration level i.The CBiand Cxiare the concentrations of the linarin and the other reference standards(X),at the concentration level i.The Fxand RRT were calculated by formulas as follows:RRFx=ABi.Cxi/AxiCBi,RRT=tx/tB,tBis the retention time of the linarin CRS obtained from a chromatogram,in minutes.tXis the retention time of the others.
2.7.Validation of the SDDMC method
The SSDMC method,with linarin selected as the internal single standard,was validated for specifcity,stability,linearity,limit of quantifcation(LOQ),precision(within-and between-day variability),and robustness.The results of the precision,accuracy and robustness calculated by the SDDMC method were compared with the results obtained from the traditional external standard method using the paired t-test.
2.8.HPLC fngerprint analysis
Data analysis was performed by a professional software named Similarity Evaluation System for Chromatographic Fingerprint of Traditional Chinese Medicine composed by Chinese Pharmacopoeia Committee(Version 2009 A),which was recommended by CFDA of China.The simulative mean chromatogram as a representative standard chromatogram for a group of chromatograms of the CIF samples from different sources was calculated and generated by this software.The correlation coeffcient of similarity between each chromatographic profle of CIF samples and the simulative mean chromatogram was calculated,respectively.
PCA is a sophisticated technique widely used for reducing the dimensions of multivariate problems.It reduces the dimensionality of the original data set by explaining the correlation among a large number of variables in terms of a smaller number of underlying factors without losing much information.In this study,the PCA was performed on the relative peak area of common peaks in the HPLC fngerprints by SPSS 19.0 software.
3.Results and discussions
3.1.Calculation of relative response factor and relative retention time
Using linarin as the internal standard,the RRFs and RRT of six standards were calculated based on results of linearity.As shown in Table 2,the RRF of luteolin-7-O-β-D-glucoside was 1.01.The RRFs of chlorogenic acid,3,4-di-O-caffeoylquinic acid,3,5-di-O-caffeoylquinic acid,and 4,5-di-O-caffeoylquinic acid were 0.71,0.65,0.61 and 0.60,respectively.The similarity of RRFs could be explained by the similar chromophore groups of analytes.In addition,the RSDs were less than 4.0%,indicating that the values of the RRFs obtained on the same instrument were stable.The calculation of RRT was necessary so that the peaks could be identifed with only the internal standard.From Table 2,the RSDs of RRT less than 0.7%suggested that the RRTs obtained on the same instrument were highly repeatable.
Through seven concentration standard solutions,the ruggedness of RRT and RRF of six analytes were compared with different equipments(Agilent 1260 and Shimadzu 20A)and columns(Phenomenex).As shown in Table 1,the average RRFs of luteolin-7-O-β-D-glucoside,chlorogenic acid,3,4-di-O-caffeoylquinic acid,3,5-di-O-caffeoylquinic acid, and 4,5-di-O-caffeoylquinic acid were 0.74,1.01,0.65,0.61, and 0.60,respectively,using the above described conditions. RSDs were<4.7%.The results showed that the RRFs for each analytes were quite similar at detection wavelength of 326 nm under different columns and HPLC instruments, indicating a good durability of the RRFs.The RRT was quite stable in different equipments and columns with the RSDs<2%.In general,both the RRT and RRF were fuctuated in a relative narrow range in different equipments and columns.
3.2.Validation of the SDDMC method
3.2.1.Specifcity and stability
The specifcity was estimated by comparing the consistency of the retention time and UV spectrum between a sample andthe corresponding reference standard.Fig.1A and B shows that the six main components in the chromatogram of CIF could be identifed by the corresponding standards.Meanwhile,peak purity detection function of PDA was used,which confrmed acceptable purity of the six analytic peaks in the sample chromatogram.
The stability of sample solution was analyzed by calculating the coeffcient of variation of the main peak area in the same sample after storage for different times.As shown in Table 3,the results of the stability evaluation indicated thatthe sample solution was stable for almost 24 h,with the RSDs of peak areas less than 3%.
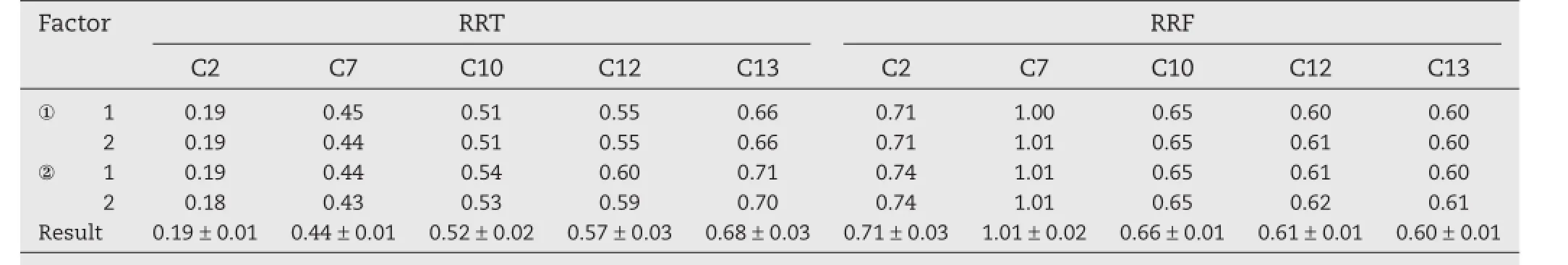
Table 1–Ruggedness of RRT and RRF of six analytes(n=6).

Table 2–The results of linearity,LOQ,RRF and RRT.

Table 3–The results of stability of CIF sample solution (n=1).

Fig.1–Representative HPLC chromatograms of (A,B)mixed standards and(C)CIFwith the compounds numbered as follows:(1)Gallic acid,(2)Chlorogenic acid, (3)Cryptochlorogenic acid,(4)Coffeic acid, (7)Luteolin-7-O-β-D-glucoside,(10)3,4-di-O-caffeoylquinic acid,(12)3,5-di-O-caffeoylquinic acid, (13)3,4-di-O-caffeoylquinic acid,(16)Linarin.
3.2.2.Linearity range
Calibration plots for the six compounds were obtained over the calibration range at seven concentration levels.The seven point calibration curves for six compounds showed a linear correlation between concentration and peak area withr>0.9997 (Table 2).The limits of quantifcation for six analytes were listed in Table 2,which indicated high sensitivity under the HPLC conditions.
3.2.3.Precision and accuracy
The within-day variability of precision was performed by three replicates at three different concentrations.The between-day variability of precision was analyzed by three replicates each day on three consecutive days.The accuracy was determined by a recovery test performed by spiking all of the reference standards into a sample(0.25 g)of CIF powder,followed by extraction and analysis as above described.Three concentration levels, covering the 75.00–125.0%ranges of a known amount of analyte in the sample,were established in order to perform the recovery test.
For establishing the within-day variability,the RSDs of six analytes were within 2.0%.Furthermore,the results performed by the two methods showed no remarkable differences using paired t-test(P=0.161).For the between-day variability,the RSDs of six analytes were within 2.2%except for linarin (RSD=4.4%).The results obtained by the two methods showed no remarkable differences(P=0.301).For the recovery test,the recoveries of all of the analytes were in the range 90.0–108% with the RSDs less than 5.4%.The recovery between the two methods showed no remarkable differences(P=0.068).Through the above discussed validation and comparison,it was shown that the SSDMC method could obtain coincident and reasonable accuracy and precision compared with the traditional external standard method.
3.2.4.Robustness
In order to apply the SSDMC method in different laboratories, the factors studied were adjusted subjectively one variable at a time(OVAT)to investigate the signifcant affecting factors for thismethod.Eightexperimentalconditionswereslightlyvaried, including column temperature(±1°C)fow rate(±0.1 ml/min), concentration of acid(±0.1%),ratio of organic phase(±1%),time of gradient(±1 min),wavelength(±2 nm),and injection volume (±5 μl).The parameters,including RRF,RRT,total content,theoretical plate number(N),and peak-tailing factor(T),were compared to determine the robustness of the SSDMC method. The results as followed:(1)the values of RRFs at each level of the eight factors were maintained at 0.71,1.01,0.65,0.61,0.60, respectively,and consistent with the results in section 3.1, showing that all the operational factors had little infuence on the RRFs.(2)The values of RRT at each level of the eight factors were well distributed except for the factors of fow rate and ratio of organic phase.RSDs ranged in 2.0%–5.4%at the two levels of factors,indicating that a slight variation of fow rate and proportion of mobile phase had a signifcant effect on RRT. (3)The total contents of the six analytes varied on the factor of fow rate(RSDs ranged in 3%–4%),but were invariable on the other factors.The phenomenon was consistent with the tendencyofN.(4)ThevaluesofTateachleveloftheeightfactors were stable.The factor of fow rate had a signifcant affect on R,but all the values ofRwere more than 2.
Above all,the fow rate,and proportion of mobile phase should be controlled in the HPLC method for CIF,while the other conditions were allowed to vary in the certain range on the basis of above analysis.
3.3.Application of the SSDMC method
The validated SSDMC and ESM methods were employed to assay 33 batches of CIF from different origins.The total contents of six analytes were listed in Table 4.From the Box plot in Fig.2,the six major components were detected in all CIF samples,whereas their contents differed greatly from each other especially for linarin(component 16),probably due to the differences of harvest time,geographical climate,process and storage.In addition,the SSDMC method could obtain accordant values compared with the traditional external standard method for assay of the different CIF samples(P=0.318).
3.4.HPLC fngerprint analysis
Based on the results of determination,HPLC fngerprints for CIF were established.Reference chromatographic fngerprint for CIF extracts was generated based on the 33 samples obtained from various sources.The pattern fngerprints of CIF were illustrated in Fig.3.As shown in Fig.1B,a good separation and reproducible chromatogram was achieved and 16 peaks were marked as the common peaks(from peak 1 to peak 16)in the chromatograms of the 33 raw herbs.Nine peaks(1,2,3,4,7, 10,12,13,16)in the HPLC fngerprint profle were identifed by comparing the UV spectra and retention time with the reference compounds.Peak 16(linarin),the most important active constituents of CIF(China Pharmacopoeia Committee 2010), was chosen to calculate the relative retention time(RRT)andrelative peak area(RPA)of all the other peaks.The results from the 33 batches of samples indicated that the RPAs of the 16 common peaks varied dramatically,but the RRT was invariable for the herb and,thus,was a suitable parameter for identifcation.

Fig.2–Box plot of content distribution of six analytes in the 33 samples of CIF.C2 was component 2.So did the others.Total was total content of the six analysis.

Table 4–Total Content(%)of investigated compounds in CIF from SSDMC and ESM method.The similarities of chromatograms of 33 batches of CIF samples.
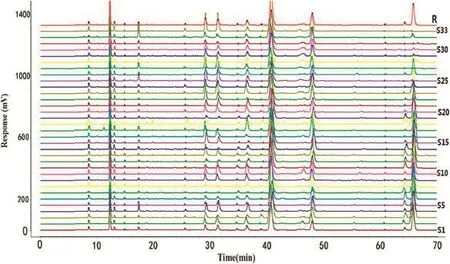
Fig.3–HPLC fngerprint of 33 batches of CIF from different origins;R:reference chromatogram generated from all CIF samples.
3.4.1.Similarity evaluation
It was necessary that chromatographic fngerprint of CIF from various sources should be evaluated by their similarities,which obtained from the calculation on the correlative coeffcient of original data.Thus the correlation coeffcient between each chromatogram of CIF samples and the simulative mean chromatogram was shown in Table 1.The results indicated that the samples shared different correlation coeffcients of similarities.Furthermore,the samples from eastern China involving Henan,Hubei,Jiangsu,Anhui,Zhejiang and Jiangxi(except S5, S6,and S7)achieved the higher value of similarity among 33 samples,which suggested that samples from the above regions shared a similar chromatographic pattern.While,the samples from southwestern and southern China including Gui zhou, Sichuan,Guangxi,Hunan,Guangdong had the adverse result, suggesting that these products differ from those with a high similarity value.
3.4.2.Principle component analysis(PCA)
Since the mean chromatograms of each source indicated that differences among samples mainly existed in the content variations of common components.In order to evaluate the discrimination ability of these common components,PCA was employed using the RPAs of 16 common peaks as input data instead of the full spectrum of fngerprints without any preprocessing.The RPAs of common constituents in 33 batches of CIF samples from various sources formed a matrix of 16×33. The frst two principal components,PC 1 and PC 2,which refected 95.5%of the total variance in these samples,were chosen to provide a convenient visual aid for identifying inhomogeneity in the data sets.As depicted in Fig.4,the samples could be classifed into three groups,which were marked as groups I–III.Regardless of the overlapped marks,group I consisted of 16 samples totally from southeastern China including Anhui, Henan,Hubei,Jiangsu,and Zhejiang except S26 from Guangdong;group II consisted of 11 samples collected from southwestern and southern China involving Sichuan,Guizhou, Guangxi,Hunan,Guangdong and Jiangxi;group III consisted of 3 samples from Anhui and Hubei.The samples classifed into the same group had similar chemical properties/components. Groups I and II were partially overlapped and the overlapping area consisted of 3 samples from Zhejiang and Jiangxi as the transitional regions of eastern and southern China.Group III was far from group I,which could be explained by the differences of storage,harvesting time or other potential factors. Additionally,the data listed in Table 1 confrmed that the similarity values of samples in groups I–III was in ft the decrement tendency.

Fig.4–The scatter plot obtained by PCA of the 33 samples of CIF.
4.Conclusions
In summary,a simple,effcient and enhanced quality assessment method for CIF was established.In contrast to the conventional quality assessment standard of CIF,the SSDMC method overcame the scarcity and cost of chemical reference substances.It is the frst time using SSDMC method for simultaneous quantifcation of the six components in CIF.The method proved to have good linearity,precision,recovery,stability and robustness.Additionally,fngerprint analysis in combination with SA and PCA was performed to identify the CIF samples from different regions under the same chromatographic conditions.The results demonstrated that HPLC coupled with determination using SSDMC method and fngerprint analysis is a powerful and practical tool for comprehensive quality control of CIF and can be replicated for other herbal medicines with multiple components.
Acknowledgement
This study was fnancially supported by Liaoning Innovative Research Team in University(LNIRT,Grant No.LT2013022).
R E F E R E N C E S
[1]Kim JE,Jun S,Song M,et al.The extract ofChrysanthemum indicumLinne inhibits EBV LMP1-induced NF-κB activation and the viability of EBV-transformed lymphoblastoid cell lines.Food Chem Toxicol 2012;50:1524–1528.
[2]Cheng WM,Li J,You TP,et al.Anti-infammatory and immunomodulatory activities of the extracts from the inforescence ofChrysanthemum indicumLinne.J Ethnopharmacol 2005;101:334–337.
[3]Zhu SY,Yang Y,Yu HD,et al.Chemical composition and antimicrobial activity of the essential oils ofChrysanthemum indicum.J Ethnopharmacol 2005;96:151–158.
[4]Yoshikawa M,Yoshikawa T,Murakami T,et al.Medicinal fowers.I.Aldose reductase inhibitors and three neweudesmane-type sesquiterpenes,kikkanols A,B,and C,from the fowers ofChrysanthemum indicumL.Chem Pharmaceut Bullt 1999;3:340–345.
[5]Shen S,Sha YF,Deng CH,et al.Quality assessment of fos chrysanthemi indici from different growing areas in China by solid-phase microextraction-gas chromatography-mass spectrometry.J Chromatogra A 2004;1047:281–287.
[6]Hao BJ,Wu YH,Wang JG,et al.Hepatoprotective and antiviral properties of isochlorogenic acid A from Laggera alata against hepatitis B virus infection.J J Ethnopharmacol 2012;144:190–194.
[7]Clifford MN,Wu W,Kirkpatrick J,et al.Profling the chlorogenic acids and other caffeic acid derivatives of herbal chrysanthemum by LC-MSn 397.J Agric Food Chem 2007;55:929–936.
[8]Cao GH,Sofca E,Priora RL.Antioxidant and prooxidant behavior of favonoids:structure-activity relationships.Free Radic Biol Med 1997;22:749–760.
[9]Zhang YL,Yang XX,Zhang XN.Quantitative analysis of astragaloside IV in traditional Chinese medicine‘Huang-Qi-SiWu’Capsules by HPLC/UV.J Chin Pharma Sci 2010;19:223–228.
[10]Cheng XM,Zhao T,Yang T,et al.HPLC fngerprints combined with principal component analysis,hierarchical cluster analysis and linear discriminant analysis for the classifcation and differentiation of Peganum sp.Indigenous to China.Phytochem Ana 2010;21:279–289.
[11]Qiao CF,Han QB,Song JZ,et al.Chemical fngerprint and quantitative analysis of Fructus Psoraleae by high-performance liquid chromatography.J Sep Sci 2007;30:813–818.
[12]Gao XY,Jiang Y,Lu JQ,et al.One single standard substance for the determination of multiple anthraquinone derivatives in rhubarb using high-performance liquid chromatography-diode array detection.J Chromatogra A 2009;1216:2118–2123.
[13]Hou RY,Jiao WT,Qian XS,et al.Effective extraction method for determination of neonicotinoid residues in tea.J Agric Food Chem 2013;61:12565–12571.
[14]Magdalena K,Tomasz C,Renata J,et al.Comprehensive two-dimensional gas chromatography for determination of the terpenes profle of blue honeysuckle berries.Food Chem 2014;152:88–93.
[15]Matteo B,Marco P,Tullia GT,et al.Analysis of green tea catechins:comparative study between HPLC and HPCE. J Agric Food Chem 2003;81:631–638.
[16]Jezussek M,Schieberle P.A new LC/MS-method for the quantitation of acrylamide based on a stable isotope dilution assay and derivatization with 2-mercaptobenzoic acid.Comparison with two GC/MS methods.J Agric Food Chem 2003;51:7866–7871.
[17]Antonio DT,Alberto F,Vincenzo F.Quantitation of acrylamide in foods by high-resolution mass spectrometry. J Agric Food Chem 2014;62:74–79.
Abbreviations:HPLC,high performance liquid chromatography;LOD,limit of detection;LOQ,limit of quantifcation;NDS,natural dietary supplement;RSD,relative standard deviation;SSDMC,a single standard to determinate multi-components;SA,similarity analysis;PCA, principal component analysis;RRF,relative response factor;RRT,relative retention time.
*
.School of Pharmacy,Shenyang Pharmaceutical University,Shenyang 110016,China.Tel.:+8602423984392;fax: +8602423986259.
E-mail address:lqyxm@hotmail.com(Q.Li).
Peer review under responsibility of Shenyang Pharmaceutical University.
http://dx.doi.org/10.1016/j.ajps.2015.08.010
1818-0876/©2016 The Authors.Production and hosting by Elsevier B.V.on behalf of Shenyang Pharmaceutical University.This is an open access article under the CC BY-NC-ND license(http://creativecommons.org/licenses/by-nc-nd/4.0/).
杂志排行

Asian Journal of Pharmacentical Sciences的其它文章
- Preparation and characterization of solidifed oleanolic acid–phospholipid complex aiming to improve the dissolution of oleanolic acid
- Development of a topical ointment of betamethasone dipropionate loaded nanostructured lipid carrier
- Supersaturation induced by Itraconazole/Soluplus®micelles provided high GI absorption in vivo
- Rapid and sensitive analysis of melatonin by LC-MS/MS and its application to pharmacokinetic study in dogs
- Effect of process parameters on the recrystallization and size control of puerarin using the supercritical fuid antisolvent process
- The 1st Euro-Mediterranean Workshop:Natural Products in Health and Diseases:Cairo,Egypt, March 2,2015