An Improved Methodology to Overcome Key Issues in Human Fecal Metagenomic DNA Extraction
2017-01-11JitendraKumarManojKumarShashankGuptaVasimAhmedManuBhambiRajeshPandeyNarSinghChauhan
Jitendra KumarManoj KumarShashank GuptaVasim Ahmed Manu BhambiRajesh PandeyNar Singh Chauhan*g
1Department of Biochemistry,Maharshi Dayanand University,Rohtak 124001,India
2Ayurgenomics Unit,Translational Research and Innovative Science ThRough Ayurgeomics,Council of Scientifc and Industrial Research,Institute of Genomics and Integrative Biology,New Delhi 110020,India
METHOD
An Improved Methodology to Overcome Key Issues in Human Fecal Metagenomic DNA Extraction
Jitendra Kumar1,a,Manoj Kumar1,b,Shashank Gupta1,c,Vasim Ahmed1,d, Manu Bhambi1,e,Rajesh Pandey2,f,Nar Singh Chauhan1,*,g
1Department of Biochemistry,Maharshi Dayanand University,Rohtak 124001,India
2Ayurgenomics Unit,Translational Research and Innovative Science ThRough Ayurgeomics,Council of Scientifc and Industrial Research,Institute of Genomics and Integrative Biology,New Delhi 110020,India
Metagenomic DNA
extraction;
Gut microbiome;
Human feces;
16S rRNA
Microbes are ubiquitously distributed in nature,and recent culture-independent studies have highlighted the signifcance of gut microbiota in human health and disease.Fecal DNA is the primary source for the majority of human gut microbiome studies.However,further improvement is needed to obtain fecal metagenomic DNA with suffcient amount and good quality but low host genomic DNA contamination.In the current study,we demonstrate a quick,robust,unbiased, and cost-effective method for the isolation of high molecular weight(>23 kb)metagenomic DNA(260/280 ratio>1.8)with a good yield(55.8±3.8 ng/mg of feces).We also confrm that there is very low human genomic DNA contamination(eubacterial:human genomic DNA marker genes=227.9:1)in the human feces.The newly-developed method robustly performs for fresh as well as stored fecal samples as demonstrated by 16S rRNA gene sequencing using 454 FLX+. Moreover,16S rRNA gene analysis indicated that compared to other DNA extraction methods tested,the fecal metagenomic DNA isolated with current methodology retains species richness
Introduction
and does not show microbial diversity biases,which is further confrmed by qPCR with a known quantity of spike-in genomes.Overall,our data highlight a protocol with a balance between quality, amount,user-friendliness,and cost effectiveness for its suitability toward usage for cultureindependent analysis of the human gut microbiome,which provides a robust solution to overcome key issues associated with fecal metagenomic DNA isolation in human gut microbiome studies.
Isolation of metagenomic DNA from fecal samples with current method
Humans live in close association with microbes which act as a constituent organ[1,2].The total number of microbes residing in the human body,especially in the gut,outnumbers that of human cells[3].A vast array of recent studies has identifed many microbial enterotypes in the human gut[4-10]and their potential roles in immunity[5,7,8],development[8],digestion [9],and other functions[10].
Majority of the studies have used 16S rRNA gene sequencing to understand the community structure,composition,and functional diversity of the human gut microbiome [1-4].The successofthese culture-independentstudies depends primarily on the quality and quantity of metagenomic DNA isolated from the given samples[11-13].Therefore,isolation of metagenomic DNA with a good quality from a heterogeneous source like human feces has been a challenging task.
Human feces are complex due to the presence of fbers, microbes,undigested particles,nucleases,and human cells [14].Removal of fbers and undigested particles from feces is diffcult,which in turn affects overall quality and quantity of metagenomic DNA being isolated[14-16].Moreover,the presence of these impurities also compromises effcient lysis of microbial cells.Many microbes elude complete lysis,resulting in an uneven contribution of metagenomic DNA and eventually compromised microbial diversity[11,17].Additionally, human genomic DNA remnants in metagenomic DNA affect the metagenomic sequence data output,thereby increasing per base sequencing cost[6].To overcome these challenges, many metagenomic DNA extraction protocols are being standardized,which include phenol/chloroform enzymatic lysis and freeze thaw[11,16-22].These kits have improved the quality of metagenomic DNA.However,great concerns remain pertaining to microbial diversity biasness and human genomic DNA contamination[12,17,20].The cost per sample,amount of sample,and associated impurities are other issues which may be improved upon.In the current study,we intend to overcome these limitations and provide a faster,robust,and economical metagenomic DNA extraction method with a good quality and quantity.
Methods
Fresh human fecal samples were collected from healthy individuals into a sterile container and stored at-86°C until use.Human ethical guidelines were followed strictly before engaging individuals for sample collection.The study has been conducted after ethical clearance from human ethics committee of Maharshi Dayanand University,Rohtak,Haryana, India.
The current methodology comprises two steps:(1)purifcation of the microbial cells from fecal impurities and(2)lysis of microbial cells to obtain metagenomic DNA with high molecular weight.
At the frst step,fresh feces(100 mg)were weighed into a sterile microcentrifuge tube for isolation of purifed microbial cells.Microbial cells were sequentially washed with normal saline solution(0.9%NaCl solution)and phosphate-buffered saline(PBS;pH 7.4).The washing steps were optimized for the recovery of a purifed bacterial pellet to obtain quality fecal metagenomic DNA for the downstream studies.In 5 sets of replicates,100 mg of feces were resuspended in 1 ml of normal saline solution by vortexing for 30 s and then centrifuged at ambient room temperature(RT)for 2 min with different speed of 1000 rpm(72×g),2000 rpm(287×g),3000 rpm(645×g), 4000 rpm(1147×g),and 5000 rpm(1792×g),respectively. The resulting supernatants were subjected to microscopic examination for the presence of fbers and insoluble impurities. Recovered supernatant was centrifuged again at 10,000 rpm (7168×g)for 1 min at ambient room temperature to collect microbial pellet for downstream processing.Microbial pellet from all replicates was subsequently washed with 1 ml of PBS(pH 7.4)for centrifugation with different speeds as described above.The resulting supernatants were subjected to centrifugation again at 10,000 rpm(7168×g)for 1 min to recover microbial pellet,which was used for metagenomic DNA isolation at the next step.
At the second step,the purifed microbial pellet was resuspended in 500 μl of lysis buffer containing 1%(w/v) cetyl trimethyl ammonium bromide(CTAB),100 mM of ethylenediaminetetraacetic acid(EDTA),1.5 M of NaCl, 100 mM of Na3PO4,and 100 mM of Tris-HCl(pH 8.0).After adding 2 μl of proteinase K(20 mg/ml),the mixture was incubated for 10 min at 37°C with gentle shaking at 100 rpm in orbital shaker incubator.Afterward,sodium dodecyl sulfate (SDS)was added with a fnal concentration of 1%and the incubation continued for another 20 min at 65°C with intermittent shaking. The lysate was centrifuged at 13,000 rpm (12,114×g) for 5 min at ambient room temperature.The resulting supernatant was collected and mixed with an equal volume of saturated phenol:chloroform: isoamylalcohol(25:24:1),which isthen subjected to centrifugation at 10,000 rpm(7168×g)for 5 min at RT.The aqueous phase was collected and metagenomic DNA was precipitated with 0.6 volume of isopropanol and pelleted by centrifugation at 13,000 rpm(12,114×g)for 5 min.After washing twice with 70%ethanol,the resulting DNA was dried and fnally dissolved into a 50 μl of 1×Tris-EDTA buffer (pH 8.0).
The qualitative and quantitative analysis of the metagenomic DNA was performed by agarose gel electrophoresis, restriction endonucleasedigestion (Sau3A1),NanoQuant (Tecan Group,Mannedorf,Switzerland)estimation,and Qubit®dsDNA HS Assay Kit(Life Technologies,Carlsbad, CA).Metagenomic DNA recovered from all replicates was compared for qualitative and quantitative parameters to obtain optimized condition for metagenomic DNA isolation from human feces.The optimized method is outlined in Figure 1 and was then used to isolate the metagenomic DNA from 10 one-month-old frozen feces stored at-86°C and 50 random fecal samples(including both fresh and frozen samples)to evaluate its robustness.
Isolation of metagenomic DNA from fecal samples with commercial methods/kits
Metagenomic DNA was isolated from fresh or frozen human fecal samples using 4 commercial kits,including Power Fecal® DNA Isolation Kit(MO BIO Laboratories,Carlsbad,CA) [11](referred as method A hereafter),Extract MasterTMFecal DNA Extraction Kit(Epicentre,Madison,WI)[21](referred as method B hereafter),Favor PrepTMStool DNA isolation Kit(Favorgen Biotech,Ping-Tung,Taiwan,China)(referred as method C hereafter),and QIAamp DNA Stool Kit (QIAGE,Hilden,Germany)(referred as method D hereafter) as instructed by the respective manufacturers.The quality and quantity of fecal metagenomic DNA were assessed as mentioned above.
qPCR amplifcation
The qPCRs were performed on a 7500 Fast Real Time PCR system (ABI,Life Technologies,Carlsbad,CA)using 2×KAPA SYBR Fast qPCR master mix(universal)from KAPA Biosystems(Wilmington,MA).The 20 μl reaction mixture contained 1 μl of metagenomic DNA/human genomic DNA(30 ng/μl),7.5 μl of 2×SYBR Green,1 μl of primer mix(forward and reverse primer of 0.5 mM),0.4 μl of master mix(High Rox),and 5.1 μl of nuclease-free water.Primers used include human MUC5B-specifc primers and eubacterial 16S rRNA gene-specifc primers(Table S1).The experiment was performed in triplicate using human genomic DNA as control.The qPCRs were performed with holding stage at 95°C for 20 s,40 cycles of 95°C for 30 s,and 60°C and fnal melt curve stage with continuous mode at 95°C for 15 s,60°C for 60 s,95°C for 15 s,and 60°C for 15 s.Melt curve analysis of the primers confrmed the high effciency of the primers andamplifed product generated during the reaction.The relative quantifcation was carried out using the 2-ΔΔCTmethod.
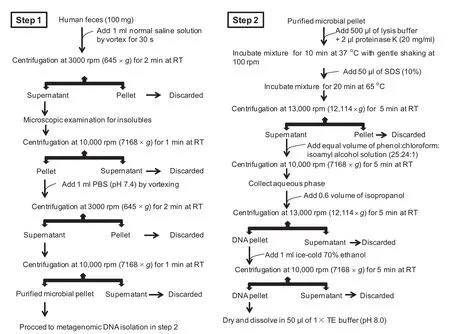
Figure 1 Workfow for fecal metagenomic DNA extraction using the current method
Effcacy of current methodology was further validated with fecal samples spiked with Escherichia coli DH10B(Invitrogen, Carlsbad,CA),Bacillus subtilis(MTCC-2057,Chandigarh, India),and Aspergillus niger(MTCC-514,Chandigarh,India). The effcacy of current methodology was analyzed by quantitatively comparing the presence of marker genes of 16S rRNA and internal transcribed spacer(ITS)in DNA extracted using current methodology from spiked stool samples and DNA extracted from respective pure cultures.All DNA quantifcation experiments were performed with host-specifc primers including 16S rRNA gene primers (16S120_FP and 16S345_RP)for microbes and ITS gene primers(ITS 1F and ITS 4B)for fungus[23]using qPCR with aforementioned PCR settings.
Pyrosequencing and sequence analysis of 16S rRNA gene
The 16S rRNA gene was amplifed from metagenomic DNAs extracted with different methodologies following optimized PCR conditions[24].The resulting amplicons were analyzed using agarose gel electrophoresis and quantifed with Qubit® dsDNA HS Assay Kit.
The amplifed 16S rRNA gene from the metagenomic DNA isolated with our methodology and with method A were also sequenced with Roche 454 GS FLX+system,following the manufacturer’srecommendations.The16S rRNA gene sequences generated and used in the current study were submitted as a NCBI Bioproject(Accession ID:PRJNA295000). Subsequently,Quantitative Insights Into Microbial Ecology (QIIME)pipeline was implemented for pyrosequencing data analysis[24],along with 16S rRNA gene sequence data obtained from the Human Microbiome Project(HMP)[25]. Variability analysis of 16S rRNA gene sequences was performed using QIIME statistical tools[24-26].
Results and discussion
Isolation of metagenomic DNA from human feces
A number of efforts to optimize a methodology for metagenomic DNA isolation from feces have been undertaken [11,13,15,20].Although progress has been made in this regard, problems of limited applicability(e.g.,microbial diversity studies only)and acceptability due to their complex process,poor metagenomic DNA quality,host genomic DNA contamination,low yield,and high cost have still left scope for a methodology to overcome the shortcomings[11,17-21].
The current methodology is a two-step process with handson time of 80-90 min.At the frst step,various large size insoluble impurities like undigested food particles and dietary fbers were removed to collect a clean translucent microbial pellet. Purifcation of microbial pellet would enable effcient lysis of microbial cells and a better DNA recovery.In the second step, the microbial cells were treated with lysis buffer and proteinase for microbial cell lysis to achieve a high yield of metagenomic DNA.The microscopic examination showed that feces washing and following centrifugation at3000 rpm (645×g) removed majority of insoluble impurities with a minimum microbial loss.While washing and following centrifugation at 1000 rpm(72×g)and 2000 rpm(287×g)enabled maximum microbial recovery with abundant insoluble impurities, washing and following centrifugation at 4000 rpm(1147×g) and 5000 rpm(1792×g)have removed all impurities with huge microbial loss.Among all replicates,feces washing and following centrifugation at 3000 rpm(645×g)yielded good quality of purifed microbial pellet,and subsequently metagenomic DNA with high molecular weight free from molecular inhibitors of comparable yield(Table S2).
The DNA yield was 55.80±3.80 ng/mg of feces(Table 1). Spectrometric analysis using NanoQuant showed 260/280 ratio of 1.83±0.02.Qualitative analysis with agarose gel electrophoresis also confrmed good integrity for DNA with size>23 kb(Figure S1)and negligible RNA presence(Figure 2A).
The robustness of the protocol was also confrmed using one-month-old frozen fecal sample stored at-86°C with a yield of 40.00±5.00 ng/mg of feces.The protocol was repeated for an independent set of 50 non-redundant fecal samples with varying texture and consistency.The quality (260/280 ratio of 1.8-1.9)and quantity(47.5±2.5 ng/mg of feces)was consistent within all replicates(Table 1).
In summary,current methodology has been successfully used to isolate the metagenomic DNA from one-month-old frozen fecal sample stored at-86°C with a minimal effect on yield and quality,which was a challenge as per reported metagenomic DNA isolation studies[13,14,20-22].
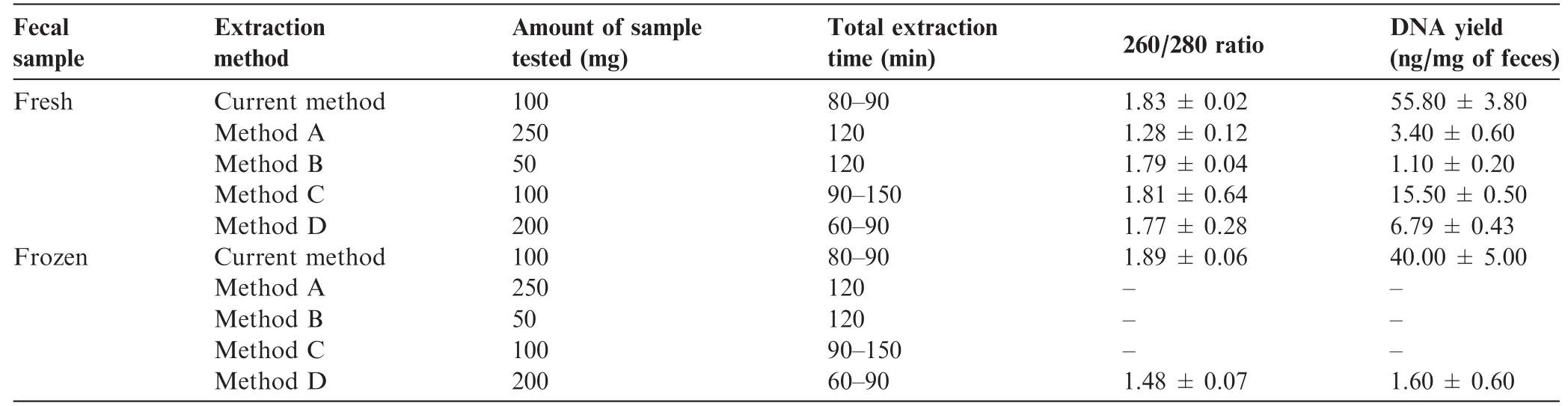
Table 1 Qualitative and quantitative analysis of human fecal metagenomic DNA extraction using different methods
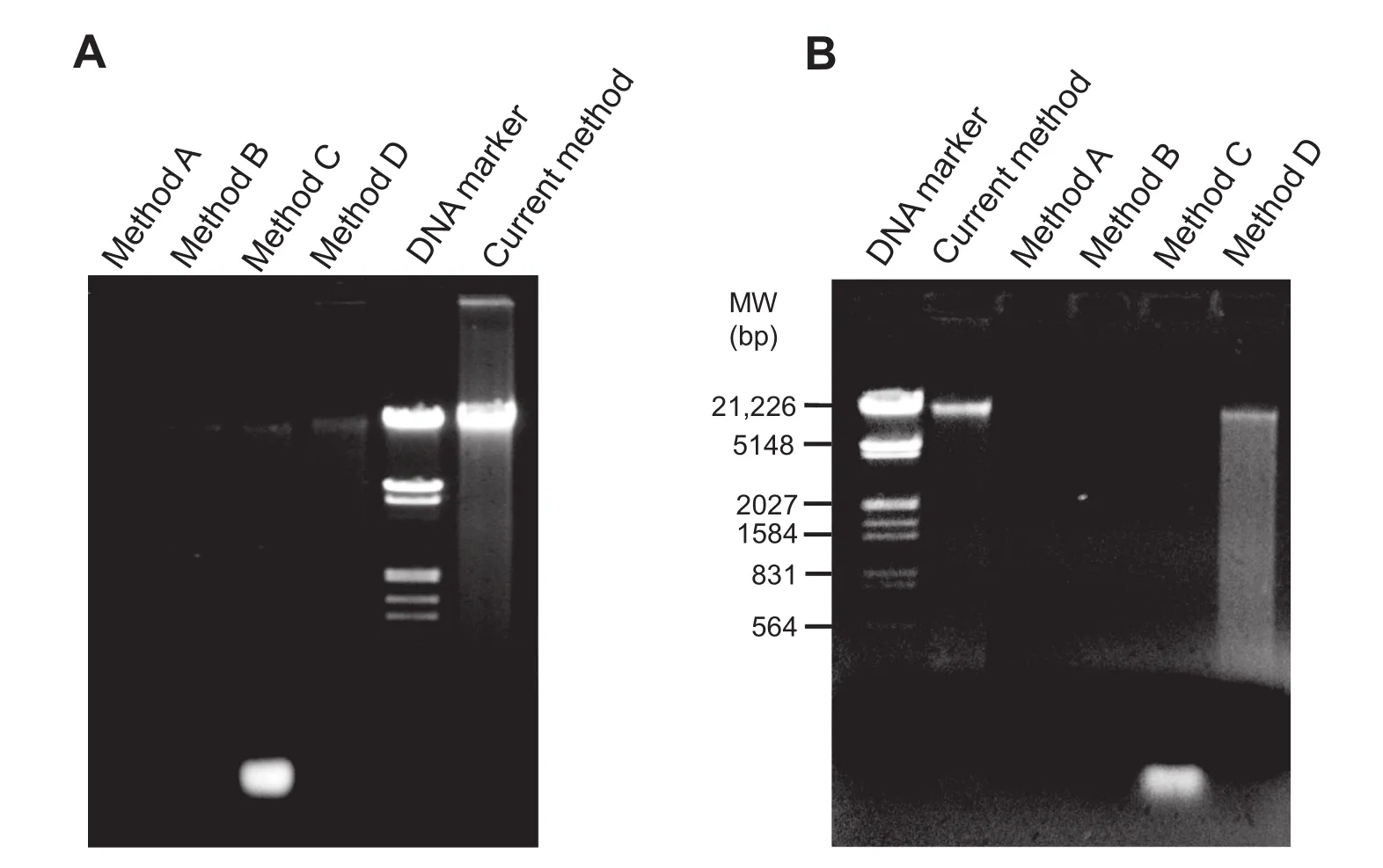
Figure 2 Gel electrophoresis of human fecal metagenomic DNA isolated with different methods
Comparative analysis with commercial methods/kits
To evaluate the relative performance of our current method,we compared it with other commercially available metagenomic DNA isolation kits for human feces[11,16,21].As instructed by kit manufacturers,various defned amount of fecal sample were used for commercial methods A,B,C,and D.Notably, low yields of DNA were obtained using these methods in comparison to that using current method(Table 1).Gel electrophoresis analysis indicated poor DNA yield with method A,B,and D,while RNA contamination was observed in DNA isolated with method C(Figure 2A).Similarly,spectrometric analysis showed a lower yield and compromised 260/280 ratios for DNA extracted using methods A-D (Table 1).Moreover,these methods did not work well for extracting metagenomic DNA from one-month-old stored fecal sample(Table 1).We failed to extract metagenomic DNA from frozen feces using methods A-C,while a low amount of DNA was recovered using method D.On the other hand,although with a reduced yield when compared to using fresh feces,more signifcant DNA with decent 260/280 ratio was recovered when using our method to extract DNA from frozen feces.In contrast,a poor quality of metagenomic DNA with negligible output was observed with one-month-old frozen feces at-86°C with all other methods tested(Figure 2B).
The metagenomic DNA isolated from fresh human fecal samples with current method and other commercial methods was further analyzed for contamination of host genomic DNA using qPCR.The amplifcation ofeubacterial and human genomic DNA marker genes,16S and MUC5B, indicated a huge difference in the copy number of eubacterial:human genes(227.9:1)in the metagenomic DNA isolated with current method.However,the eubacterial:human ratios were compromised for the fecal metagenomic DNA isolated using commercial methods(213:1 for method A,212:1 for method B,and 20.143:1 for method C,220.8:1 for method D). Similar observation on host genomic DNA contamination in fecal metagenomic DNA was recorded during the HMP study using method A[6].
The cycle threshold(Ct)value is used for absolute copy number quantifcation of a target gene in qPCR.In general, lower Ct value indicates higher copy number of target gene, while higher Ct value means low copy number of target gene. A low Ct value of 6.780±0.231 was observed for eubacterial 16S,and of 34.740±0.374 for human-specifc MUC5B using metagenomic DNA isolated with current method.Metagenomic DNA isolated with other methods showed varied Ct values:19.310±0.185(method A),22.010±0.089(method B),28.130±0.821(method C),and 11.780±0.295(method D)for 16S gene,respectively,while Ct values of 32.690 ±0.332(method A),34.090±0.166(method B),28.270 ±3.426(method C),and 32.630±0.647(method D)were observed forhuman-specifcMUC5B gene,respectively. Lower Ct values of other methods for human-specifc MUC5B gene compared to our method indicated few human DNA remnants in the metagenomic DNA isolated with current protocol, refecting superior representation of the eubacterial-specifc metagenomic DNA in comparison to the limited presence of human genomic DNA in metagenomic DNA extracted with current methodology.
qPCR analysis of known genome spike-in experiments was further performed for validation of the effcacy and unbiased cellular lysis of current methodology.qPCR analysis indicated a good recovery of 79.85±12.16%for E.coli DH10B genomic DNA in fecal samples spiked with E.coli DH10B and 72.09±5.02%for B.subtilis genomic DNA in fecal samples spiked with B.subtilis.However,a slightly lower recovery (36.35±9.49%)was observed for A.niger genomic DNA in fecal samples spiked with A.niger.A good amplifcation observed with the metagenomic DNA isolated using current method indicates the fecal metagenomic DNA isolated with current methodology was free from the impurities that could interfere the reaction[17,19,21].
Evaluation of current method for human microbiome studies by pyrosequencing
To further evaluate the applicability of the isolated DNA samples for sequencing analysis,the 16S rRNA gene(V1-V4 region)was amplifed from metagenomic DNA isolated using our protocol and method A after column purifcation.These amplicons were subjected to sequencing analysis using Roche 454 FLX+.As a result,we obtained 57,689 sequencing reads with an average read length of~530 bp for the 4 samples tested.These sequences were quality fltered(>Q30)to remove ambiguous and chimeric sequences.Finally,54,262 highquality reads were retained for the following downstream analysis.
As shown in Table 2,more 16S rRNA gene sequencing reads were recovered for DNA isolated using method A than using current method.The reads were processed using QIIME de novo clustering pipeline to get the operational taxonomic units(OTUs).Similarly,we found that more OTUs were detected for DNA isolated using method A than using current method(Table 2).We then estimated the species richness by normalizing the read counts with the sequence value(-e) based on the minimum number of high quality sequencing reads for each sample,which is 6300 in the current study, and analyzed the total alpha diversity.Our results showed that despite fewer reads,more species were observed for DNA isolated using current method than using method A.An average number of 611 and 635 microbial species were observed in samples H1 and H2,respectively,which were isolated using the current method,in comparison to 515 and 496 microbial species from the same source feces with DNA isolated using method A(Table 2).In the meantime,we also noticed a higher Shannon diversity index.Taken together,these data indicate that even for the same fecal samples,sequencing outcomes can be greatly affected by the metagenomic DNA isolation methods used[12].
Number of observed species relative to the increasing number of 16S rRNA gene sequencing reads obtained was also analyzed to obtain the identifcation rate of new OTUs (Figure 3A).The identifcation rate of microbial phylotypes was slightly higher for 16S sequencing reads generated from metagenomic DNA extracted with current method than method A.These results highlight the usefulness of the current method to capture species richness even from lower number of sequencing reads.More observed species with better identifcation rate can be obtained from metagenomic DNA isolated using current methodology(Figure 3A).
Biasness has been reported between microbial diversity of a host sample and its metagenomic DNA extraction method [12,22].To test whether there exist such biases in current methodology,a β diversity analysis was performed between the microbial diversities obtained from DNA isolated with current method,HMP data,and method A.The β diversities show positioning of the samples based on their microbial diversities.Principal coordinate analysis(PCoA)plots were generated from 16S rRNA gene sequencing reads(Figure 3B). Compared to method A,signifcant variability of interindividual microbial diversity was observed for the current method.It indicates that the current methodology performed better with respect to unbiased lysis of microbial cells and contribution into the total metagenomic DNA pool.
Overall,the results we presented here highlight the effciency of the current protocol to achieve better yield and quality of the gut metagenomic DNA while simultaneously retaining the sample enrichment with respect to the bacterial species present in the human gut.The current protocol has not been tested for other host species.Different host species are of specifc diet pattern and life style,which in turn affect the constituents of the feces.Therefore,the protocol presented here may warrant specifc but minor adjustments to account for species-specifc gut metagenomic DNA isolation.For most of the species,the requirement may be met by maneuvering the relative concentration of NaCl and the centrifugation conditions which are important for purifying microbial cells from the background impurities.Given the user-friendliness of the current protocol,this may be optimized at individual level without great diffculties.
Authors’contributions
JK,MK,and VA performed sample collection,DNA isolation,and experiment optimization.RP performed real-timePCR analysis and 16S rRNA gene sequencing.SG carried out sequence analysis.MB and NSC designed the study and drafted the manuscript.All authors read and approved the fnal manuscript.

Table 2 Microbial diversity analysis of the 16S rRNA gene sequencing reads

Figure 3 Diversity analyses of 16S rRNA gene sequencing reads
Competing interests
The authors have declared that there are no competing interests.
Acknowledgments
This study was supported by the Council of Scientifc and Industrial Research(CSIR)-sponsored research scheme[Grant No.60(0099)/11/EMRII]and the Major Research Project of the University Grants Commission(UGC)(Grant No.41-1256/2012 SR),India.MB and MK were supported by the fellowships,respectively,from the Department of Biotechnology (DBT)and the Department of Science and Technology(DST), India.
Supplementary material
Supplementary material associated with this article can be found,in the online version,at http://dx.doi.org/10.1016/j. gpb.2016.06.002.
[1]Bik EM.Composition and function of the human-associated microbiota.Nutr Rev 2009;67:S164-71.
[2]Eckburg PB,Bik EM,Bernstein CN,Purdom E,Dethlefsen L, Sargent M,et al.Diversity of the human intestinal microbial fora. Science 2005;308:1635-8.
[3]Fierer N,Ferrenberg S,Flores GE,Gonzalez A,Kueneman J. From animalcules to an ecosystem:application of ecological concepts to the human microbiome.Annu Rev Ecol Evol Syst 2012;43:137-55.
[4]Gill SR,Pop M,Deboy RT,Eckburg PB,Turnbaugh PJ. Metagenomic analysis of the human distal gut microbiome. Science 2006;312:1355-9.
[5]Human Microbiome Project Consortium.Structure,function and diversity of the healthy human microbiome. Nature 2012;486:207-14.
[6]Human Microbiome Project Consortium.A framework for human microbiome research.Nature 2012;486:215-21.
[7]Robinson CJ,Bohannan BJ,Young VB.From structure to function:the ecology of host-associated microbial communities. Microbiol Mol Biol Rev 2010;74:453-76.
[8]Spor A,Koren O,Ley R.Unravelling the effects of the environment and host genotype on the gut microbiome.Nat Rev Microbiol 2011;9:279-90.
[9]Zoetendal EG,Vaughan EE,de Vos WM.A microbial world within us.Mol Microbiol 2006;59:1639-50.
[10]Mandal RS,Saha S,Das S.Metagenomic surveys of gut microbiota.Genomics Proteomics Bioinformatics 2015;13:148-58.
[11]Andersen AW,Bahl MI,Carvalho V,Kristiansen K,Ponten TS, Gupta R,et al.Choice of bacterial DNA extraction method from fecal material infuences community structure as evaluated by metagenomic analysis.Microbiome 2014;2:2-19.
[12]Goodrich JK,Di Rienzi SC,Poole AC,Koren O,Walters WA, Caporaso JG,et al.Conducting a microbiome study.Cell 2014;158:250-62.
[13]Yuan S,Cohen DB,Ravel J,Abdo Z,Forney LJ.Evaluation of methods for the extraction and purifcation of DNA from the human microbiome.PLoS One 2012;7:e33865.
[14]McOrist AL,Jackson M,Bird AR.A comparison of fve methods for extraction of bacterial DNA from human faecal samples.J Microbiol Methods 2002;50:131-9.
[15]Monteiro L,Bonnemaison D,Vekris A,Petry KG,Bonnet J, Vidal R,et al.Complex polysaccharides as PCR inhibitors in feces:Helicobacter pylori model.J Clin Microbiol 1997;35:995-8.
[16]Yu Z,Morrison M.Improved extraction of PCR-quality community DNA from digesta and fecal samples.Biotechniques 2004;36:808-12.
[17]Henderson G,Cox F,Kittelmann S,Miri VH,Zethof M,Noel SJ, et al.Effect of DNA extraction methods and sampling techniques on the apparent structure of cow and sheep rumen microbial communities.PLoS One 2013;8:e74787.
[18]Clement BG,Kitts CL.Isolating PCR-quality DNA from human feces with a soil DNA kit.Biotechniques 2000;28:640-6.
[19]Li M,Gong J,Cottrill M,Yu H,de Lange C,Burton J,et al. Evaluation of QIAGEN®DNA Stool Mini Kit for ecological studies of gut microbiota.J Microbiol Methods 2003;54:13-20.
[20]Nechvatal JM,Ram JL,Basson MD,Namprachan P,Niec SR, Badsha KZ,et al.Fecal collection,ambient preservation,and DNA extraction for PCR amplifcation of bacterial and human markers from human feces.J Microbiol Methods 2008;72:124-32.
[21]Thornton CG,Passen S.Inhibition of PCR amplifcation by phytic acid,and treatment of bovine fecal specimens with phytase to reduce inhibition.J Microbiol Methods 2004;59:43-52.
[22]Kuczynski J,Lauber CL,Walters WA,Parfrey LW,Clemente JC, Gevers D,et al.Experimental and analytical tools for studying the human microbiome.Nat Rev Genet 2012;13:47-58.
[23]Gardes M,Bruns TD.ITS primers with enhanced specifcity for basidiomycetes-application to the identifcation of mycorrhizae and rusts.Mol Ecol 1993;2:113-8.
[24]Morowitz MJ,Denef VJ,Costello EK,Thomas BC,Poroyko V, Relman DA,et al.Strain-resolved community genomic analysis of gut microbial colonization in a premature infant.Proc Natl Acad Sci U S A 2011;108:1128-33.
[25]Caporaso JG,Kuczynski J,Stombaugh J,Bittinger K,Bushman FD,Costello EK,et al.QIIME allows analysis of high-throughput community sequencing data.Nat Methods 2010;7:335-6.
[26]Edgar RC,Haas BJ,Clemente JC,Quince C,Knight R. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 2011;27:2194-200.
Received 4 January 2016;revised 7 June 2016;accepted 15 June 2016 Available online 23 November 2016
Handled by Kang Ning
*Corresponding author.
E-mail:nschauhan@mdurohtak.ac.in(Chauhan NS).
aORCID:0000-0003-4222-446X.
bORCID:0000-0002-6872-4975.
cORCID:0000-0001-8839-7003.
dORCID:0000-0002-1360-6023.
eORCID:0000-0002-5276-6548.
fORCID:0000-0002-4404-8327.
gORCID:0000-0003-4546-9358.
Peer review under responsibility of Beijing Institute of Genomics,Chinese Academy of Sciences and Genetics Society of China.
http://dx.doi.org/10.1016/j.gpb.2016.06.002 1672-0229©2016 The Authors.Production and hosting by Elsevier B.V.on behalf of Beijing Institute of Genomics,Chinese Academy of Sciences and Genetics Society of China.
This is an open access article under the CC BY license(http://creativecommons.org/licenses/by/4.0/).
杂志排行

Genomics,Proteomics & Bioinformatics的其它文章
- Reading and Interpreting the Histone Acylation Code
- Biological Databases for Hematology Research
- T Cell Repertoire Diversity Is Decreased in Type 1 Diabetes Patients
- Identifcation of Risk Pathways and Functional Modules for Coronary Artery Disease Based on Genome-wide SNP Data
- Plant Proteins Are Smaller Because They Are Encoded by Fewer Exons than Animal Proteins
- Announcement of Distinguished GPB Articles 2012-2015