基因表达谱的非参缺失森林填补算法研究*
2017-01-10第三军医大学军事预防医学院卫生统计学教研室400038
第三军医大学军事预防医学院卫生统计学教研室(400038)
吴小姣 李高明 易大莉 刘 岭 张彦琦 易 东 伍亚舟△
·方法介绍·
基因表达谱的非参缺失森林填补算法研究*
第三军医大学军事预防医学院卫生统计学教研室(400038)
吴小姣 李高明 易大莉 刘 岭 张彦琦 易 东 伍亚舟△
目的评估不同基因表达谱数据集下,多种算法在缺失数据中的填补效果,并分析其对无监督基因表达谱聚类的影响。方法在不同缺失比例的情况下,分别采用非参缺失森林填补法、贝叶斯线性回归法、蒙特卡洛多重填补法和K邻近填补法进行填补,通过均方根误差(NRMSE)和聚类准确率(F值)两个指标来评估不同方法的填补效能和聚类效果,并用模拟数据集进行测试和乳腺癌数据集进行验证。结果随着缺失比例的增加,四种填补方法的NRMSE都逐渐上升;任意缺失比例下,相比于其他三种方法非参缺失森林填补法的填补优势明显。缺失比例为5%、10%、20%和30%的乳腺癌数据集,非参缺失森林填补法的NRMSE依次为0.1951(95%CI,0.1945~0.1953)、0.2776(95%CI,0.2783~2791)、0.4003(95%CI,0.3986~0.4002)和0.4974(95%CI,0.4658~0.5104);聚类效果的准确率为1.0、0.91、0.88和0.82。结论非参缺失森林填补算法实现简单,对数据集的要求较低,比传统填补算法具有更好的稳定性和精确度,可保留较多的基因信息供后续的功能聚类等分析。
基因表达谱 缺失数据 缺失森林法 聚类
生物医学研究中基因表达微阵列是一种强有力的工具,但现存的很多分析方法都要求微阵列的数据是完整的。由于存在多种原因,如不充分的实验方案,图像损坏,芯片上的灰尘或划痕等,使得实际上获得的数据阵列通常是有缺失的,这在一定程度上影响了数据后续分析结果的准确性和可靠性,如差异表达基因的筛选、基因功能聚类、基因调控网络建立和生物标志物检测等。目前芯片缺失数据填补估计方法的文献较多,可以大致分为四类:(1)局部算法:K邻近距离法[1]、局部最小二乘法[2]等;(2)全局算法:奇异值分解法[3]、贝叶斯填补算法[4]等;(3)混合算法:linC-mb[5];(4)利用生物信息辅助算法:POCS[6]、HAI填补[7]等。这些填补方法大多属于参数统计方法,且都要求数据集的分布已知。实际上,基因表达谱数据集往往具有复杂的数据结构且无任何先验知识,非参数模型方法对此却能取得很好的效果;同时针对不同数据集的不同分析目的,将多种方法同时进行比较的文献较少,其研究尚有较大空间。本文介绍的非参缺失森林填补算法即为一种非参数统计方法,首次将其应用于基因表达谱缺失数据的填补,并将其与常用的几种填补方法(如贝叶斯线性回归法[4]、蒙特卡洛多重填补法[8]和K邻近填补法[2])的填补效果进行比较,最后分析各种填补方法对无监督基因表达谱聚类的影响,为同类研究提供方法学借鉴。
理论与算法
1.非参缺失森林的填补方法
随机森林算法[9]要求应变量是完整的,才能训练出森林,Stekhoven在此基础上进行改进,提出了缺失森林算法[10],它可以直接用已观测到的完整部分数据集训练出的随机森林来预测缺失值,而不依赖于应变量的完整性。
假定数据集X=(X1,X2,…,Xp)是一个N×P维的矩阵(N个基因,P个样本),将其中任意一个可能含缺失数据的变量记为XS。应变量和自变量的观测值、缺失数据分别记为Yobs、Ymis和Xobs、Xmis。
具体的填补步骤如下:首先,用均数或其他填补方法对X的所有缺失值作初步的猜测,并将变量XS按缺失值的数量升序排列,令这个初步填补后的矩阵为Xold。对每一个变量XS,缺失森林算法的填补过程为:
(1)首先用应变量Yobs和自变量Xobs拟合一个随机森林;
(2)然后将Xmis作为特征变量输入,用训练后的随机森林来预测缺失数据Ymis,令新预测填补后得到的矩阵为Xnew;
(3)重复此填补过程,直到符合停止标准γ,即新填补的数据矩阵Xnew和前一个数据矩阵Xold的差值首次开始增加时;连续变量N间的差值定义为
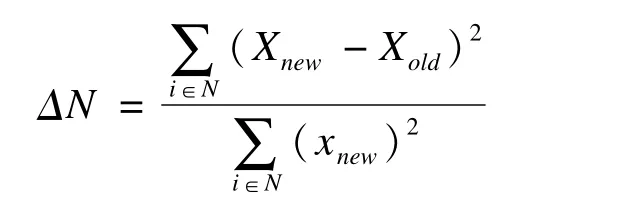
2.缺失填补的效果评价
任何一种填补方法,都有各自的优点和缺点。通常情况下,采用标准化均方根误差(normalized root mean square error,NRMSE)作为不同方法填补效果的评价指标[11]:

其中Ri为估计值,Ii为原始值,Std(Ii)为原始值的标准差。NRMSE的值越小表示其估计的越准确、性能越好,反之结果越差。
3.聚类的效能评价
用层次聚类法对四种方法填补的完整数据集进行聚类,并对聚类结果进行评价。层次聚类法产生一个嵌套聚类的层次,算法最多包含N步,在第t步执行的操作就是在前t-1步的聚类基础上生成新聚类。假定对N个对象进行聚类,层次聚类法[12-14]过程如下:
(1)初始时共有N类,每个类有一个对象构成。令序号m=0,l(m)=0。
(2)在D中寻找最小距离d[r,s]=min d[(i),(j)]。
(3)将两个类r和类s合并成一个新类(R,S),令m=m+1,L(m)=d[r,s]。
(4)更新距离矩阵D:将表示类r和类s的行列删除,同时加入表示新类(r,s)的行列;同时定义新类(r,s)与旧类(k)的距离为d[(k),(r,s)]=min(d[(k),(r)],d[(k),(s)])。
(5)重复(2)~(4)步,直到所有对象合并成一个类为止。
在聚类的过程中,每次抽取一个填补缺失数据的方法为检验样本,以完整数据集的样本构成训练集,用训练集训练分类器,然后对检验样本进行检验,分别记录下每个支持向量机在检验样本阳性类和阴性类的真阳性数(TP),真阴性数(TN),假阳性数(FP),假阴性数(FN)。一般用F值[15]方法对聚类的效果进行评价:

其中,P=TP/(TP+FP);R=TP/(TP+FN);β为伪错误的概率。F值越大表示其聚类效能越好,反之越差。
数据集
1.数据来源
本实验采用两个基因表达数据集,第一个使用R软件的ARTIVA包模拟一个多元正态分布的表达谱数据集,表示1024个基因在15个实验水平下的不同表达。第二个数据集来自GEO数据库上公开发表的乳腺癌基因表达谱数据[16],该数据集为6365个基因,15个实验样本,两个数据集都为非时间序列型结构。
2.统计分析
分别对模拟和乳腺癌数据集,采用统计软件包R3.2.4编程,按照一定百分比(如5%、10%、20%、30%)产生随机性缺失数据,在统计软件R下分别使用非参缺失森林法、贝叶斯线性回归法、蒙特卡洛多重填补法和K邻近法对缺失的乳腺癌表达谱数据集进行填补,并进行基因功能聚类分析的效果评估。需要加载的程序包有:affy、compositions、mice、missForest、impute、hclust、cutree。
结 果
1.基于均方根的填补效果评价
四种算法的填补效果如图1所示。无论使用哪种填补方法,NRMSE的值都会随着缺失比例的增加而逐渐上升。如乳腺癌数据集在缺失比例为10%时,非参缺失森林法、蒙特卡洛多重填补法、K邻近填补法和贝叶斯线性回归法的NRMSE依次为0.2671、0.3202、0.3190和0.4115。在任意缺失比例下,非参缺失森林填补算法的优势较明显。

图1 不同填补方法在不同缺失比例下的填补效果(NRMSE值)
在不同的缺失比例下,用非参缺失森林填补法对不同缺失比例下的模拟数据集填补10次,均方根误差的标准差和置信区间见表1,在5%、10%、20%和30%的缺失比例下,其均方根误差的标准差分别为0.0006、0.0006、0.0016和0.0312,置信区间的宽度分别为0.0008、0.008、0.0016和0.0446,说明该算法的稳定性强、精确度高。
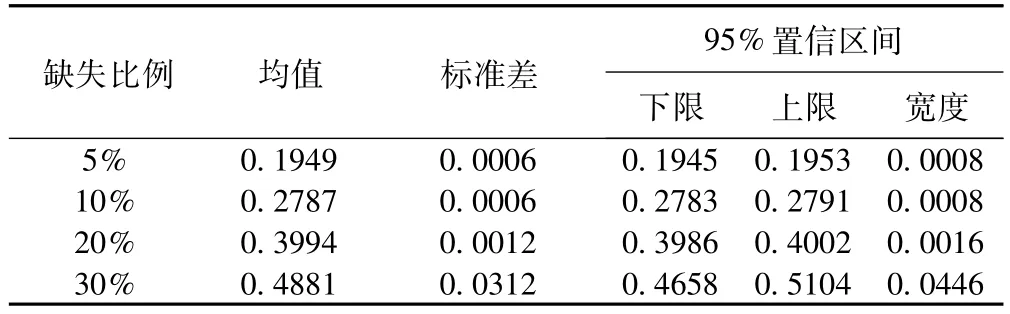
表1 不同缺失比例下NRMSE均值及标准差(填补10次时)
2.基于聚类分析的效果评价
图2为四种填补方法在两个数据集中不同缺失比例下基因功能聚类分析的准确率(F值)。在不同缺失比例下,不同填补方法对数据集的聚类效果有较大的影响;填补方法上,使用非参缺失森林算法填补数据集的聚类效果优于其他三种算法。在5%缺失比例的时候,所有填补方法的F值都高于0.93,聚类效果好;乳腺癌数据集在20%缺失比例的时候,非参缺失森林法、K邻近填补法、贝叶斯线性回归法和蒙特卡洛多重填补法的F值依次为0.8819、0.8717、0.7934和0.7501,整体趋势上和模拟数据集中的聚类效果一致。
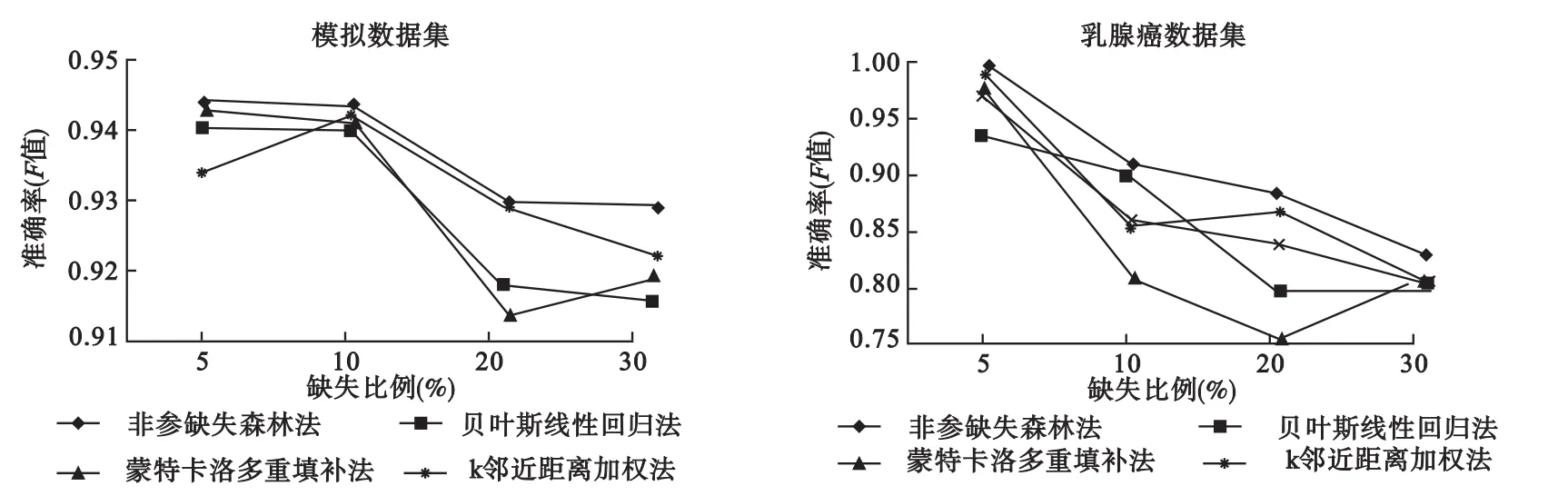
图2 不同填补方法在不同缺失比例下基因功能聚类分析的准确率(F值)
讨 论
本文采用不同的方法对含有缺失值的不同数据集进行填补,并应用于后续的基因功能聚类分析,通过NRMSE和聚类效果(F值)来评价各种填补方法的优劣及其适用性,不仅发展和丰富了基因表达谱缺失数据的填补模型方法,而且为基因表达谱数据分析技术提供了生物信息学方法方面的指导。
尽管在不同的数据集上依据不同的指标对各缺失值处理方法进行评价,结论会有细微的差别,但总体来看,随着缺失比例的增加,基于非参缺失森林的填补方法优势逐渐显现,它既提高了缺失估计的精度和稳定性,又可以保留较多的基因信息供后续的功能分析且具有很高的聚类效能。在缺失比例比较小的情况下,蒙特卡洛多重填补法和K邻近距离加权法的填补效果也比较好;从聚类结果的准确率来看,如果运用不恰当的填补方法会对后续表达谱的研究起误导性作用,但是直接对含有缺失数据的乳腺癌数据进行聚类,效果不理想,这也从侧面说明了根据缺失数据集特点选择正确填补方法的重要性。
本文介绍的非参缺失森林填补方法具有良好的应用前景,它对数据集的结构要求较低、实现简单,相比于传统填补算法具有更好的稳定性和准确度,可以保留较多的基因信息供后续的功能聚类等分析目的。有关缺失森林程序包的更多扩展功能参见missForest程序包说明。本研究结果是基于较大样本量且只用于表达谱数据的聚类分析目的,将其推广到小样本数据和其他分析目的(如差异表达基因筛选和基因调控网络建立等),可能会受到一定限制,我们将继续进行后续的分析与探讨。总之,本文通过不同填补方法的研究,为基因表达谱数据缺失填补策略的建立和缺失填补方法对基因表达谱后续不同分析目的生物学影响及其程度的评估,打下了坚实的理论和实践基础。
[1]Nanni L,M ing J,Du Y,et al.M issing value imputation for gene expression data:computational techniques to recovermissing data from available information.American Journal of Medical Genetics,2011,12(5):498-513.
[2]Troyanskaya O,Cantor M,Sherlock G,etal.M issing value estimation methods for DNA m icroarrays.Bioinformatics,2001,17(6):520-525.
[3]Kim H,Golub G.M issing value estimation for DNA m icroarray gene expression data:local least squares imputation.Bioinformatics,2005,21(2):187-198.
[4]Oba S,Sato M,Takemasa I,et al.A Bayesian m issing value estimation method for gene expression profile data.Bioinformatics,2003,volume 19(16):2088-2096.
[5]Jörnsten R,Wang H,Welsh W,et al.DNA m icroarray data imputation and significance analysis of differential expression.Bioinformatics,2005,21(22):4155-4161.
[6]Guo X,Alan W,Hong Y.M icroarray m issing data imputation based on a set theoretic framework and biological know ledge.Nucleic Acids Research,2006,34(5):1608-1619.
[7]Bai F,Liu H.M issing value imputation for m icroarray gene expression data using histone acetylation information.Smart Sensors&Sensing Technology,2008,9(1):1-17.
[8]武瑞仙,邓子兵,谯治蛟,等.利用Monte Carlo技术模拟研究不同缺失值处理方法对完全随机缺失数据的处理效果.中国卫生统计,2015(3):534-536.
[9]沈琳,胡国清,陈立章,等.缺失森林算法在缺失值填补中的应用.中国卫生统计,2014(5):774-776.[10]Stekhoven D,Bühlmann P.M issForest-non-parametric m issing value imputation for m ixed-type data.Bioinformatics,2012,28(1):112-118.
[11]Hapfelmeier A,Hothorn T,Riediger C,et al.M ice:multivariate imputation by chained equations in R.International Journal of Biostatistics,2014,45(2):1-67.
[12]刘熙,王崇骏,叶亮,等.基于最大频繁项集的层次聚类方法.广西师范大学学报(自然科学版),2009,27(3):105-108.
[13]康茜,李德玉,王素格,等.传播过程中信号缺失的层次聚类社区发现算法.计算机工程与应用,2015(9):201-206.
[14]黄健斌,康剑梅,齐俊杰,等.一种基于同步动力学模型的层次聚类方法.中国科学(信息科学),2013(05):599-610.
[15]杨燕,靳蕃,KAMEL M.聚类有效性评价综述.计算机应用研究,2008,25(6):1630-1632.
[16]Gene expression data in estrogen receptor alpha positive breast tumors with and without PIK3CA mutations[http://www.ncbi.nlm.nih.gov/bioproject/PRJNA128895.
(责任编辑:刘 壮)
国家自然科学基金项目(81273178,81573254)
△通信作者:伍亚舟,E-mail:asiawu5@sina.com
