气质联用法测定皮革中DMFo、DMAc和NMP三种残留溶剂的不确定度评定
2017-01-03杜英英邵玉婉周丽佳费国平
杜英英,邵玉婉,周丽佳,费国平
(上海市质量监督检验技术研究院,上海201114)
气质联用法测定皮革中DMFo、DMAc和NMP三种残留溶剂的不确定度评定
杜英英,邵玉婉,周丽佳,费国平
(上海市质量监督检验技术研究院,上海201114)
利用气质联用仪建立了同时测定皮革中DMFo、DMAc和NMP三种溶剂残留量的检测方法,以甲醇为溶剂,60℃超声提取60 min,GC-MS测定,外标法定量。对检测过程中不确定度的来源进行了分析,并对不确定度各个分量进行评定、合成,给出了该方法测量结果的合成不确定度和扩展不确定度,通过不确定度评定保证了检测方法的有效性。
皮革;不确定度;测定;残留溶剂;N,N-二甲基甲酰胺;N,N-二甲基乙酰胺;N-甲基吡咯烷酮
1 前言
N,N-二甲基甲酰胺(DMFo)、N,N- 二 甲 基 乙 酰 胺(DMAc)和N-甲基吡咯烷酮(NMP)是重要的化工原料和良好的溶剂。DMFo在某些皮革(如涂层皮革)或聚氨酯合成革加工过程中作为溶剂直接使用,由于环境的压力,虽然皮革在实际生产涂饰系统已经由以前的溶剂系统发展成为水基系统,但是就当前技术而言,要完全满足像高档革等的高物性要求,纯水基系统还有难度,因此涂饰剂或多或少含有有机溶剂,涂饰过程中不可避免会有少部分有机溶剂残留[1],DMFo是皮革可挥发性有机物的主要组成部分。
GB/T 29292-2012《鞋类鞋类和鞋类部件中存在的限量物质》[2]将涂层皮革和PU-TPU弹性体中的N,N-二甲基甲酰胺列为第4类限量物质,即高度怀疑对穿着者有危害的物质,但标准中未给出相应的检测方法。HJ 507-2009《环境标志产品技术要求皮革与合成革》[3]中要求挥发性有机物≤100 mg/kg,其中的挥发性有机物包含了DMFo。DMFo、DMAc和NMP均已被列入REACH法规高度关注物质(SVHC)清单。
为了保障人们健康安全并与相关标准法规要求相适应,在目前国内外检测标准尚不齐全或不完善的情况下,有必要对皮革材料中残留有机溶剂的检测方法进行研究,并建立一种切实可行的检测方法。本文利用自行建立的检测方法对皮革中DMFo、DMAc和NMP三种残留溶剂含量进行检测,并基于JJF 1059-1999《测量不确定度评定与表示》[4]的一般要求对检测方法的不确定度进行评定,通过不确定度评定保证了检测方法的有效性。

表1 DMFo、DMAc和NMP的保留时间及特征离子
2 试验部分
2.1 仪器与试剂
Thermo Trace DSQ-Ⅱ气相色谱-质谱联用仪(赛默飞世尔科技(中国)有限公司);BL6-180C超声波清洗机(上海比朗仪器有限公司);BSA224S-CW分析天平(赛多利斯科学仪器(北京)有限公司);可调移液器(Eppendorf中国有限公司)。
标准物质:N,N-二甲基甲酰胺(DMFo)(气相色谱纯,纯度99.99%,上海沃凯化学试剂有限公司)、N,N-二甲基乙酰胺(DMAc)标准品(纯度99.5%,Dr.Ehrenstorfer)、N-甲基吡咯烷酮(NMP)标准品(纯度99.5%,Dr.Ehrenstorfer)。
甲醇(色谱纯,赛默飞世尔科技(中国)有限公司)。
2.2 检测原理和分析步骤
在超声波作用下,用甲醇萃取试样中的残留溶剂,气相色谱-质谱仪(GC-MS)测定,外标法定量。具体步骤如下:称取约1.0 g(精确至0.0001 g)试样于带螺纹口的试剂瓶中,加入10 mL甲醇,60℃下超声萃取60 min,吸取少量试液用有机滤膜过滤,供GC-MS分析测试。
2.3 GC-MS仪器分析条件
气相色谱条件:色谱柱为毛细管柱(DB-Wax,30 m×0.25 mm × 0.25 μm),载气为纯度≥99.999%的高纯氦,流量为1.0 mL/min,进样方式为不分流进样,进样量为1 μL,进样口温度为250℃,以20℃/min的速率从55℃升温至240℃,保持2 min。
质谱条件:离子源温度为230℃,四极杆温度为150℃,离子源为EI源,电子能量70 eV,接口温度为250℃。DMFo、DMAc和NMP的保留时间及特征离子[5]见表1。
2.4 标准溶液的配制与稀释
2.4.1 DMFo、DMAc和NMP标准储备溶液
准确称取DMFo、DMAc和NMP标准品各10 mg,用色谱纯甲醇溶解并分别定容于10 mL容量瓶中,得到1 000 mg/L的标准储备溶液,密封保存在4℃冰箱中,有效期1年。
2.4.2 DMFo、DMAc和NMP混合标准中间液
移取0.5 mL的质量浓度为1 000 mg/LDMFo、DMAc和NMP标准储备液至25 mL容量瓶中,用甲醇定容至刻度,得到20 mg/L的混合标准中间液,密封保存在4℃冰箱中,有效期一个月。
2.4.3 标准曲线工作溶液
分别取0.125 mL、0.5 mL、1.25 mL、2.5 mL和5 mL质量浓度为 20 mg/L的 DMFo、DMAc和NMP标准中间液,用甲醇定容于10 mL容量瓶中,得到质量浓度分别为0.25 mg/L、1 mg/L、2.5 mg/L、5 mg/L和 10 mg/L标准工作溶液,与20 mg/L标准溶液组成六个不同浓度的标准曲线工作溶液。
2.5 测试结果计算公式
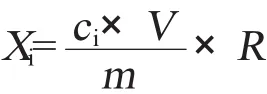
其中Xi——样品中残留溶剂的含量,mg/kg;
ci——由校准曲线得到的样液中残留溶剂的浓度,mg/L;
V——样液定容体积,mL;
m——样品质量,g;
R——回收率。
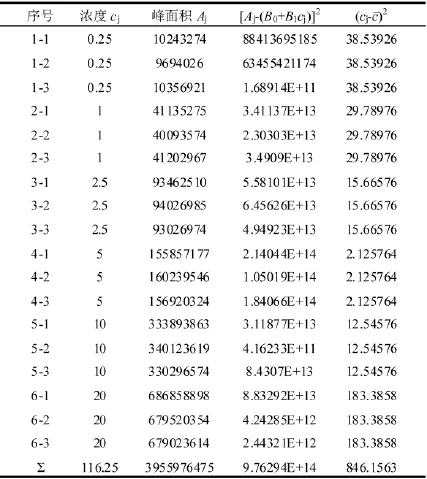
表2 DMFo校准曲线各校准点的浓度及对应的峰面积值
3 不确定度来源的确定
由测试结果计算公式可以看出,皮革中DMFo、DMAc和NMP三种残留溶剂测定的不确定度主要来源于以下五个方面:
(1)校准曲线引入的不确定度
(2)标准溶液配制引入的不确定度
(3)样品称量引入的不确定度
(4)样品定容体积引入的不确定度
(5)样品重复测量引入的不确定度
(6)方法回收率引入的不确定度
4 不确定度来源的量化
4.1 校准曲线引入的不确定度
使用校准曲线计算DMFo、DMAc和NMP残留溶剂的含量,以DMFo为例对校准曲线引入的不确定进行分析。用20 mg/L混合标准中间液配制6个不同浓度的标准曲线工作溶液,质量浓度分别为:0.25 mg/L、1 mg/L、2.5 mg/L、5 mg/L、10 mg/L和20 mg/L。六个标准溶液分别被测量三次,以DMFo为例对校准曲线引入的不确定进行分析,DMFo仪器的响应值结果见表2:
校准曲线为:Aj=B1·cj+B0
其中,Aj——标准溶液的峰面积;
cj——标准溶液的浓度,mg/L;
B1——校准曲线斜率;
B0——校准曲线截距。
以最小二乘法拟合曲线,得到的线性方程为:Aj=33798207.98cj+1496377.60,线性相关系数R2=0.9990。即B1=33798207.98,B0=1496377.60。
样品溶液测定2次,由校准曲线得到样品溶液中DMFo的平均浓度c0=9.750 mg/L。以最小二乘拟合法建立校准曲线产生的不确定度计算公式如下:

其中,p——样品溶液的测定次数,p=2;
n——标准溶液的测定次数,n=18;
c0——样品溶液中DMFo的质量浓度,c0=9.750 mg/L;
DMAc和NMP由校准曲线引入相对不确定度的分析方法与DMFo相同,在此就不再赘述,仅给出计算结果,DMAc和NMP校准曲线引入的相对不确定度分别为:0.022和0.019。
4.2 标准溶液配制引入的不确定度ur(cs)
以位于校准曲线中间的浓度点10 mg/L对标准溶液配制引入的不确定度进行评估。根据校准曲线的配制方法,首先称取10 mg标准物质,定容至10 mL容量瓶,得到质量浓度为1 000 mg/L的标准溶液储备液。用可调移液器(量程为1 mL)移取0.5 mL储备液至25 mL容量瓶中,得到20 mg/L的标准溶液中间液,用可调移液器(量程为10 mL)移取5 mL中间液至10 mL容量瓶中,得到10 mg/L的标准溶液,其浓度cs可用下式表示:

由上式可知,该标准溶液的不确定度由四个分量组成:
4.2.1 标准物质的纯度引入的不确定度ur(std)
由标准物质证书,标准物质DMFo的纯度为99.99%,因此其相对不确定 度 ur(std)=
DMAc的纯度为99.5%,因此其相对不确定度。ur(std)=
NMP的纯度为99.5%,因此其相对不确定度。ur(std)=
4.2.2 标准物质称量引入的不确定度ur(m0)
用万分之一天平称取10.0 mg DMFo,根据天平检定证书,偏差为0.1 mg。称量的标准不确定度需计算两次,一次为皮质量,一次为总质量,假设为矩形分布,则由天平最大允许误差引入的相对标准不确定度ur(m0)=
4.2.3 配制标准储备液定容引入的不确定度ur(V0)
定容体积为10 mL,根据检定证书,10 mL容量瓶的偏差为0.002 mL,假设为矩形分布,则其相对不确定度为=0.00012。定容过程中温度变化的范围为±5℃,有机液体的体积膨胀系数为1.0×10-3℃-1,假设为矩形分布,其相对不确定度为
因此标准溶液储备液配制中由定容体积V0引入的相对不确定度为:
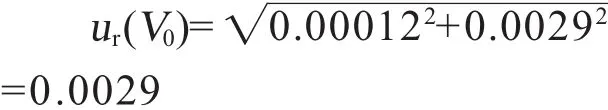
4.2.4 逐级稀释得到浓度为 10 mg/L标准工作液引入的稀释不确定度ur(D)
稀释所引入的相对标准不确定度可用下式表示:ur(D)=
根据检定证书,量程为1 mL的移液器在0.5 mL时的偏差为0.002 mL,量程为10 mL的移液器在 5 mL时的偏差为 0.02 mL,10 mL和25 mL容量瓶的偏差分别 0.002 mL和 0.005 mL,假设为矩形分布,则各体积分量 V0.5mL、V5mL、V10mL和 V25mL的相对不确定度分别为:

温度效应引入的不确定度:温度变化的范围为±5℃,有机液体的体积膨胀系数为1.0× 10-3℃-1,假设为矩形分布,则各体 积 分 量 V0.5mL、V5mL、V10mL和V25mL由温度效应导致的相对不确定度均为
通过合成,各体积分量V0.5mL、V5mL、V10mL和 V25mL的相对不确定度分别为 0.0037、0.0037、0.0029和0.0029。
因此,由逐级稀释引入的相对 不确 定 度为 ur(D)=

综上所述,DMFo标准溶液配制引入的相对不确定度为:ur(cs)

DMAc标准溶液配制引入的相对标准不确定度为:

NMP标准溶液配制引入的相对标准不确定度为:
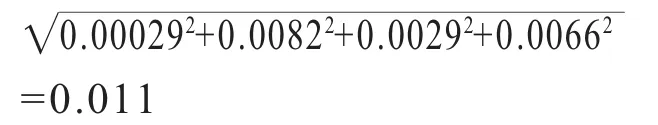
4.3 样品质量引入的不确定度ur(m)
样品称样量为1.0 g,由检定证书可知,偏差为0.2 mg,称量的标准不确定度需计算两次,一次为皮重,一次为总重,假设为均匀分布,其相对标准不确定度为
4.4 样品定容体积引入的不确定度ur(V)
前处理中,用移液枪(量程10 mL)加入10 mL甲醇进行超声提取,提取后吸取少量萃取液,用于GC-MS分析。因此,样品定容体积引入的不确定度是由移液枪带来的,由检定证书,量程为10 mL移液枪在10 mL时的偏差为0.03 mL,假设为矩形分布,其相对不确定度为检测过程中温度变化范围为±5℃,有机液体的体积膨胀系数为1.0×10-3℃-1,假设为矩形分布,其相对不确定度为
因此由样品溶液的定容体积V引入的相对不确定度为:
4.5 样品重复测量引入的不确定度ur(rep)
对样品重复测定6次,检测结果见表3。
测量重复性引入的标准不确定度由6次测量结果的平均值的标准偏差表示,因此DMFo测量重复性引入的标准不确定度为mg/kg,则测量重复性的相对标准不确定度
同样的方法计算得到,DMAc和NMP由测量重复性的相对标准不确定度分别为0.016和0.015。
4.6 方法回收率引入的不确定度ur(R)
测量方法的回收率主要来源于样品在超声萃取、转移过程中的损失及测量时可能产生的干扰。在待测阴性样品中加入一定已知浓度的标准样品,测试后计算回收率,加标量SK为100 mg/kg,结果见表3,DMFo六次测量的平均值=96.7 mg/kg,标准偏差s=3.63 mg/kg,自由度为5。标准溶液的相对不确定度为0.011。回收率(R)=测量值加 标 量 (cSK) =96.7/100=0.967。
在回收率使用中,还应采用t检验法检验回收率与1.0之间是否有显著性差异[6]。
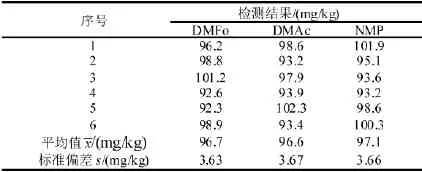
表3 样品中DMFo、DMAc和NMP含量的测定

表4 DMFo、DMAc和NMP的相对不确定度分量、不确定度值及结果表示
在95%置信区间下,当t值大于或等于t分布临界值时,则回收率与1.0之间有显著性差异需要引入修正结果;当t值小于t分布临界值时,无需进行回收率校正。在95%置信区间下,自由度为5的t分布临界值t0.95(5)=2.57,即t<t0.95(5)。因此,回收率与1.0之间无显著性差异,无需对测量结果进行回收率校正。
同样的方法计算得到DMAc和NMP方法回收率引入的不确定度分别为0.018和0.019,经过t检验法检验,两者的回收率与1.0之间均无显著性差异,也无需对测量结果进行回收率校正。
5 合成标准不确定度和扩展不确定度
皮革中DMFo、DMAc和NMP三种溶剂残留量的检测过程的不确定来源于六个分量,分别是:(1)校准曲线ur(c0);(2)标准溶液配制ur(cs);(3)样品称量ur(m);(4)样品定容体积ur(V);(5)样品重复测量ur(rep);(6)方法回收率ur(R)。
因此相对合成不确定度ur=
根据前文的分析,检测结果的各不确定度分量及大小见表4。
样品中DMFo的含量为96.7 mg/kg,则其标准不确定度u=96.7×0.032=3.09 mg/kg。
根据经验,95%置信概率下取包含因子为2,则DMFo检测结果的扩展不确定度为:U=3.09× 2=6.2 mg/kg,其测量结果及不确定度可表示为:U=(96.7±6.2) mg/kg,k=2。
同样的方法计算得到DMAc检测结果及不确定度可表示为:U= (96.6±6.8)mg/kg,k=2;NMP检测结果及不确定度可表示为:U=(97.1±6.2)mg/kg,k=2。
6 结论
本文通过对皮革中DMFo、DMAc和NMP三种残留溶剂进行测试,发现对于测定结果96.7 mg/kg、96.6 mg/kg、97.1 mg/kg,其不确定度的结果可分别表示为 :(96.7±6.2)mg/kg,k=2;(96.6±6.8)mg/kg, k=2 和(97.1±6.4)mg/kg,k=2。
分析了测量过程中不确定的来源,并对各不确定度分量进行量化,结果发现不同分量对测量不确定度的贡献值不同,其中校准曲线、样品重复测量、标准溶液配制以及方法回收率是贡献较大的不确定度分量,而样品的称量和定容体积产生的不确定度较小。这一结论,对于检测工作具有一定的指导意义,检测中应尽量提高校准曲线的拟合度,购买纯度高的标准物质并提高标准物质称量的准确性。同时在检测中应注意质量控制,可通过重复测量以及加标回收等方式保证检测的可靠性。
[1]李正军,王伟,罗永娥,等.制革生态问题及其相关皮革化学品研究评述[J].皮革科学与工程,2004,14(4):25-32.
[2]GB/T 29292-2012鞋类鞋类和鞋类部件中存在的限量物质[S].北京:中国标准出版社,2012.
[3]HJ 507-2009环境标志产品技术要求皮革与合成革[S].北京:中国标准出版社,2009.
[4]JJF 1059.1-2012测量不确定度评定与表示[S].北京:中国计量出版社,2000.
[5]杜英英,邵玉婉,赵霞,等.纺织品DMFo,DMAc和NMP残留量的GC-MS测定 [J].印染,2014,40(14):42-45.
[6]倪晓丽.化学分析测量不确定度评定指南[M].北京:中国计量出版社,2008.
Uncertainty estimation in determination of DMFo,DMAc and NMP in Leather by GC-MS
DU Ying-ying,SHAO Yu-wan,ZHOU Li-jia,FEI Guo-ping
(Shanghai Institute of Quality Inspection and Technical Research,Shanghai 201114,China)
A method for simultaneous determination of residual N,N-dimethyl formamide (DMFo),N,N-dimethylacetamide (DMAc)and N-Methyl pyrrolidone (NMP)in leather by gas chromatography mass spectrography (GC-MS)is established.Residual solvents DMFo,DMAc and NMP are ultrasonically extracted at 60oC for 60 min,using methanol as the extraction solvent,followed by the analysis of GC-MS. The concentration of each residual solvent is calibrated by external standard method.The source of uncertainty in the whole process of the measurement is analyzed.Each component of uncertainty is estimated and composed.The combined and expanded uncertainties are given.The validity of this method is evaluated with the uncertainty estimation.
leather;uncertainty;testing;residual solvent;N,N-dimethyl formamide;N,N-dimethylac etamide;N-Methyl pyrrolidone
TS 57
A
1671-1602(2016)23-0031-06
上海市质量技术监督局科研项目(2015-45)
杜英英(1984-),女,浙江台州人,博士,高级工程师,研究方向:轻工产品中有毒有害物质的分析研究。E-mail:duyy@sqi.org.cn。
